
Abstract
Background and Objective
Lenvatinib is a multikinase inhibitor that inhibits enzyme activity but induces gene expression of cytochrome P450 3A4 (CYP3A4), an important enzyme for drug metabolism. We evaluated the impact of lenvatinib on CYP3A4 using midazolam as a probe substrate in patients with advanced solid tumors. The primary objective was to determine the pharmacokinetic effects of lenvatinib on midazolam, and the secondary objective was to assess the safety of lenvatinib.
Methods
This multicenter, open-label, nonrandomized, phase 1 study involved patients with advanced cancer that progressed after treatment with approved therapies or for which no standard therapies were available.
Results
Compared with baseline, coadministration of lenvatinib decreased the geometric mean ratio of the area under the concentration–time curve for midazolam on day 1 to 0.914 (90% confidence interval [CI] 0.850–0.983) but increased it on day 14 to 1.148 (90% CI 0.938–1.404). Coadministration of lenvatinib also decreased the geometric mean ratio of the maximum observed concentration for midazolam on day 1 to 0.862 (90% CI 0.753–0.988) but increased it on day 14 to 1.027 (90% CI 0.852–1.238). There was little change in the terminal elimination phase half-life of midazolam when administered with lenvatinib. The most common treatment-related adverse events were hypertension (20.0%), fatigue (16.7%), and diarrhea (10.0%).
Conclusions
Coadministration of lenvatinib had no clinically relevant effect on the pharmacokinetics of midazolam, a CYP3A4 substrate. The adverse events were consistent with the known safety profile of lenvatinib, and no new safety concerns were identified.
ClinicalTrials.Gov Identifier
NCT02686164.
Similar content being viewed by others


Coadministration of lenvatinib with midazolam appears to have no clinically relevant effects on the pharmacokinetics of midazolam, a cytochrome P450 3A4 substrate. |
Treatment-emergent and treatment-related adverse events were manageable and consistent with the known safety profile of lenvatinib. |
1 Introduction
Lenvatinib is an oral multikinase inhibitor targeting vascular endothelial growth factor receptors 1–3, fibroblast growth factor receptors 1–4, platelet-derived growth factor receptor α, and receptor tyrosine kinases RET and KIT [1,2,3,4]. Lenvatinib is currently approved as a second-line treatment of renal cell carcinoma in combination with everolimus in the USA, for patients with progressive, radioiodine-refractory differentiated thyroid cancer (USA and EU), as a first-line treatment of unresectable hepatocellular carcinoma (USA and EU), and in combination with pembrolizumab, for the treatment of patients with advanced endometrial carcinoma that is not microsatellite instability-high or mismatch-repair deficient, who have disease progression following prior systemic therapy, and are not candidates for curative surgery or radiation (USA) [5, 6]. Lenvatinib is being assessed as a potential treatment for lung cancer [7, 8].
Cytochrome P450 3A4 (CYP3A4) is an important enzyme for drug metabolism that is found in gastrointestinal and liver tissues [9] and is responsible for the metabolism of many drugs [10]. Lenvatinib is known as a weak inducer and time-dependent inhibitor of CYP3A4 (Eisai, data on file). Midazolam is a short-acting benzodiazepine (used for sedation [11]) and is metabolized by CYP3A4 in the liver to 1′-hydroxy-midazolam (1′-OH midazolam) [12, 13]. Midazolam has been validated as a probe in other studies investigating CYP3A4 activity [14,15,16,17].
As a known CYP3A4 substrate, midazolam was used to support the recommendation that lenvatinib could be given with other drugs metabolized by the same pathway. A physiologically based pharmacokinetic model of this interaction was published previously [18], in which lenvatinib was shown to have negligible effects on midazolam metabolism. To confirm the model predictions, we conducted a phase 1 open-label study to evaluate the potential effects of lenvatinib on the induction and inhibition of CYP3A4 using midazolam as a probe substrate in patients with advanced solid tumors.
The primary objective of the study was to determine the effect of lenvatinib on CYP3A4 activity by using midazolam as a probe. The secondary objective of the study was to assess the safety of lenvatinib in this cohort of patients with advanced solid tumors.
2 Methods
2.1 Patients
Eligible patients were adults aged ≥ 18 years with a histologically or cytologically confirmed diagnosis of advanced solid tumors that had progressed following standard therapy or for which no standard therapy existed. Patients with radioiodine-refractory differentiated thyroid cancer were also eligible. Patients had to have adequate liver, bone marrow, blood coagulation, renal, and cardiac function. Patients could not be pregnant or lactating. Additional inclusion criteria were an Eastern Cooperative Oncology Group performance status of 0 to 1; adequately controlled blood pressure (defined as ≤ 150/90 mmHg at screening) with or without antihypertensive medications, and no change in antihypertensive medications within 1 week prior to onset of the lenvatinib dosing period; and any prior therapy-related toxicities being resolved to grade 0 or 1 by study entry per Common Terminology Criteria for Adverse Events (version 4.03).
Exclusion criteria comprised patients with hepatocellular carcinoma, anaplastic thyroid carcinoma with major blood vessel invasion, leptomeningeal metastases, or untreated brain metastases; urine protein levels of ≥ 1 g/24 h; patients taking medications known as potent CYP3A4 inducers or inhibitors; and patients who had previously taken lenvatinib.
Studies were conducted in accordance with the World Medical Association Declaration of Helsinki, the International Conference on Harmonisation of Pharmaceuticals for Human Use, the United States Code of Federal Regulations, the institutional review boards of the University of Pennsylvania (FWA 00004028) and the University of Texas, San Antonio (FWA 00005928), the Western Institutional Review Board, and the Biomedical Research Alliance of New York, LLC. (FWA 00003433), as well as any other applicable regulatory authorities. Informed consent was obtained from all individual patients included in the study.
2.2 Study Design and Treatment
This was a multicenter, open-label, nonrandomized, phase 1 study in patients with advanced cancer (solid tumors). Patients received lenvatinib (24 mg) orally once daily in 28-day cycles. Following the 14-day pharmacokinetic analysis presented here, patients could continue on in the extension phase. The patients received 3 single doses of midazolam (4 mg) orally on day −3 (D –3) (baseline), day 1 (D1), and day 14 (D14) (Fig. 1). D –3 was designated as the baseline evaluation point for midazolam, D1 was the evaluation point for midazolam with a single dose of lenvatinib, and D14 was the evaluation point for midazolam with lenvatinib at steady state. The variability of midazolam and magnitude of the effect considered relevant to detect a 25% difference in the mean area under the curve (AUC) between midazolam administered alone versus in combination with lenvatinib were used to calculate an ideal study size of approximately 18 patients.

Treatment schedule. aThe treatment phase for this pharmacokinetic analysis spanned day −3 to day 14. bThe extension phase started on day 15 for all patients remaining in the study
2.3 Pharmacokinetics and Safety Assessments
To achieve the primary objective, we assessed exposure to midazolam and its metabolite 1′-OH midazolam when midazolam was administered alone or with lenvatinib. Exposure was measured by using the pharmacokinetic parameters of area under the concentration–time curve from time 0 to 24 h post dose (AUC0–24) and the maximum (peak) observed drug concentration (Cmax). Additional parameters measured were the time at which the maximum (peak) plasma concentration was observed (tmax) and the terminal elimination phase half-life (t1/2). The apparent oral clearance (CL/F) and the apparent volume of distribution (Vz/F) were measured only for midazolam. Eisai internal data have shown that the population mean value for lenvatinib apparent clearance was independent of dose and time, making a noncompartmental analysis appropriate for this study.
Per the US Food and Drug Administration (FDA) [19] and the European Medicines Agency (EMA) [20] guidance documents, the effect of lenvatinib on CYP3A4 activity was defined based on its effect on mean oral clearance of midazolam and 1′-OH midazolam. A mild inhibitor is characterized by a 1.25-fold to 2-fold increase in plasma AUC. If the geometric mean ratio of AUC was > 1 but < 1.25, then the inhibition of CYP3A4 by lenvatinib would not be considered clinically significant. This assessment was performed comparing baseline (D –3) to steady state (D14).
Safety was assessed by monitoring and recording all adverse events (AEs), treatment-emergent AEs (TEAEs), treatment-related TEAEs, and serious AEs (SAEs).
2.4 Bioanalytical Assays
Blood samples (2 mL each) in K2EDTA were collected for the assessment of the pharmacokinetics of midazolam and its metabolite 1′-OH midazolam. The concentrations of midazolam and its metabolite in plasma were determined by protein precipitation followed by analyte quantification using high-performance liquid chromatography (HPLC)–mass spectrometry (AB Sciex API 4000) operated under multiple reaction monitoring positive ion mode. The HPLC system used a Luna C8(2) 5 µm, 50 × 2.00 mm column, with a gradient flow of water with 10 mM ammonium formate, pH 4.0 (Mobile Phase A) and methanol:acetonitrile (1:1, v/v) (Mobile Phase B) at a rate of 0.6 mL/min. The column was set to ambient temperature. The assay monitored m/z (mass-to-charge ratio) for midazolam (m/z 326.0 → m/z 291.2) and 1′-OH midazolam (m/z 342.0 → m/z 203.1), and for their internal standards, midazolam-D4 maleate (m/z 333.0 → m/z 295.3) and 1′-OH midazolam-D4 (m/z 346.1 → m/z 203.2), respectively. The lower limit of quantification of the assay was 0.100 ng/mL for midazolam and its metabolite, with the linear calibration ranging from 0.100–20.0 ng/mL. Appropriate bioanalytical noninterference of coadministered lenvatinib was demonstrated before the study-sample analysis. The validated method had an inter- and intra-day precision and accuracy of < 6%, with incurred sample re-analysis passing the criteria in the study-sample analysis.
2.5 Statistical Methods and Assessments
The pharmacokinetic analysis included all patients who had sufficient pharmacokinetic data to derive at least 1 pharmacokinetic parameter. The safety analysis included all patients who received at least 1 dose of midazolam or lenvatinib and had at least 1 post-dose safety assessment. Pharmacokinetic parameters were derived by noncompartmental analysis using Phoenix WinNonlin software (version 6.2 or higher) according to the standard working practices for noncompartmental pharmacokinetic analysis [21]. Plasma midazolam concentrations less than the lower limit of quantitation (0.100 ng/mL) were set to ‘missing’ in WinNonlin. To evaluate potential drug–drug interactions, the primary pharmacokinetic parameters (AUC0–24 and Cmax) for midazolam (and 1′-OH midazolam) were logarithmically transformed and used as the dependent variable, exploring treatment day as a fixed effect and patient as a random effect. The ratios (test value/reference value) of geometric means of AUC0–24 and Cmax and 2-sided 90% confidence intervals (CIs) for the ratios of these parameters were estimated.
Safety data included AEs, clinical laboratory tests, vital sign measurements, and electrocardiogram findings. TEAEs were summarized and reported as the number of patients with TEAEs by system/organ/class and Medical Dictionary for Regulatory Activities preferred term. A patient was counted only once within a system/organ/class and preferred term, even if they experienced > 1 TEAE within a specific system/organ/class and preferred term. Shifts in toxicity grade were assessed from baseline Common Terminology Criteria for Adverse Events (version 4.03) grade to maximum (worst) grade on D14. Patients were counted only once in the ‘high’ and in the ‘low’ categories, as applicable. Statistical analyses were performed using SAS version 9.3.
3 Results
3.1 Patient Disposition
A total of 30 patients were included in this study. Fifteen patients were male and most were white (19/30). The median age was 60 years (minimum, maximum: 27, 84) (Table 1).
All patients received the reference dose of midazolam, 29 patients received at least 1 dose of lenvatinib, and 19 patients received all 14 doses of lenvatinib (Fig. 2). All patients (n = 30) were included in the safety analysis set, and 29 patients were included in the pharmacokinetic analysis set. Only the 19 patients who received all 14 doses of lenvatinib were included in the comparison analysis of lenvatinib at steady state versus previous timepoints.

Patient disposition
3.2 Pharmacokinetics of midazolam with lenvatinib
A summary of the pharmacokinetic parameters for midazolam and 1′-OH midazolam can be found in Table 2. Compared with D –3, the coadministration of lenvatinib decreased the geometric mean ratio (GMR) of the AUC for midazolam on D1 but increased the GMR of the AUC on D14 (Table 3 and Fig. 3). The coadministration of lenvatinib increased the GMR of the AUC of 1′-OH midazolam on D1 and increased the GMR of the AUC on D14 (Table 3 and Fig. 3).

Forest plot of drug–drug interactions of midazolam and 1′-OH midazolam coadministered with single and multiple doses of lenvatinib in the pharmacokinetic analysis set (n = 29). D −3 midazolam only, D1 1 dose of lenvatinib and midazolam, D14 multiple doses of lenvatinib and 1 dose of midazolam, AUC0–24 area under the concentration–time curve from time 0 to 24 h after midazolam dose, D# day number, Cmax maximum observed (peak) drug concentration
Compared with D –3, the coadministration of lenvatinib decreased the GMR of Cmax for midazolam on D1, but nonsignificantly increased the GMR of Cmax on D14 (Table 3 and Fig. 3). Compared to D –3, the coadministration of lenvatinib did not produce these effects on the GMR for Cmax of 1′-OH midazolam on D1 or D14 (Table 3 and Fig. 3).
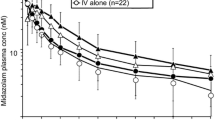
For midazolam, median tmax (minimum, maximum) was 0.75 h (0.25, 2.02) on D –3; 0.63 h (0.25, 6.00) on D1; and 0.75 h (0.25, 1.58) on D14 (Table 2). There was no statistical difference in median tmax between D –3 (0.00 h; 90% CI −0.23–0.25) and D1 or D14 (0.00 h; 90% CI −0.25–0.25) for midazolam. For 1′-OH midazolam, median (minimum, maximum) tmax was 0.75 h (0.25, 2.02) on D –3; 0.75 h (0.25, 2.00) on D1; and 0.77 h (0.25, 1.50) on D14 (Table 2). There was no statistical difference in median tmax between D –3 and D1 (0.00 h; 90% CI −0.25–0.25) or D14 (0.00 h; 90% CI −0.25–0.25) for 1′-OH midazolam. There was no statistically significant change in the t1/2 of midazolam and 1′-OH midazolam when administered with lenvatinib (Table 2). There was no statistically significant change in the CL/F and Vz/F of midazolam over time (Table 2). Coadministration of lenvatinib did not have a significant effect on the mean plasma concentration–time profiles of midazolam and 1′-OH midazolam (Table 4, Fig. 4).

Semi-log plot of mean plasma concentration–time profile of midazolam (a) and 1′-OH midazolam (b) with and without lenvatinib in the pharmacokinetic analysis (n = 29) set. D −3 day −3 (midazolam dose only), D1 day 1 (1 dose of lenvatinib and midazolam), D14 day 14 (multiple doses of lenvatinib and 1 dose of midazolam)
3.3 Safety
Most patients (n = 26 [86.7%]) had at least 1 TEAE in the treatment phase, with 10 (30.0%) experiencing an SAE (Table 5). The most frequently reported TEAEs (occurring in ≥ 20% of patients) were fatigue (26.7%), diarrhea (23.3%), and hypertension (23.3%) (Table 5). The most common treatment-related TEAEs were hypertension (20.0%), fatigue (16.7%), and diarrhea (10.0%) (Table 5). Twelve patients (40%) experienced TEAEs leading to lenvatinib dose modification. Four patients (13.3%) underwent a dose reduction and eight patients (26.7%) experienced lenvatinib treatment interruption. Two patients (6.7%) discontinued treatment due to a TEAE (one from cerebral hemorrhage and one from a Clostridium difficile infection and rectal hemorrhage). One patient died during the treatment phase due to a grade 5 TEAE (intestinal obstruction) that was determined to be related to the patient’s underlying colon cancer. The most frequently reported SAE was hypertension (n = 3 [10%]), followed by abdominal pain (n = 2 [6.7%]) (Table 5). Four patients had SAEs that were considered to be treatment related—one patient had hypertension; one patient had bradycardia and hypertension; one patient had stress cardiomyopathy and hypertension; and one had severe upper abdominal pain. All treatment-related SAEs were ≤ grade 3. No new AEs were seen in the extension phase (Eisai, data on file).
4 Discussion
This study demonstrates the lack of a statistically significant drug–drug interaction between lenvatinib and CYP3A4 substrates, which has crucial clinical implications because of the large number of drugs that are metabolized by CYP3A4. The data presented here are consistent with prior in vitro, modeling, and drug–drug interaction data.
Eisai internal data have shown that in human liver microsomes, lenvatinib showed time-dependent inhibition of midazolam 1′hydroxylation with a KI of 72.3 µmol/L. The highest Cmax seen in this study was 28.3 ng/mL, which converts to a maximum free concentration of 0.07 µmol/L. This is more than 100 times less than the in vitro KI, suggesting minimal inhibitory effects of lenvatinib upon midazolam metabolism in vivo.
Modeling data for the interaction between lenvatinib and midazolam that were presented at the American Society for Clinical Pharmacology and Therapeutics annual meeting in 2018 suggested that there is negligible drug–drug interaction [18]. A physiologically based pharmacokinetic model was used to run 10 simulated trials, each with 10 patients. The output showed that the GMR of the AUC for the concomitant use of lenvatinib plus midazolam (2 mg) remained steady across the trials for both 24 mg (GMR AUC: 1.18; 95% CI 1.16–1.20) and 32 mg (GMR AUC: 1.22; 95% CI 1.20–1.24) lenvatinib. In addition, there was no significant difference in the GMR AUC between the 2 dose levels [18]. These data correspond with the current study; when lenvatinib (24 mg) was at steady state (day 14), the GMR of the AUC for midazolam was 1.15 (90% CI 0.94–1.40). The upper limit of the 90% CI of the GMR of the AUC was beyond the upper limit of 1.25, which was likely due to the small sample.
While this study investigated the implications of lenvatinib given in combination with a CYP3A4 substrate, other studies have assessed lenvatinib in conjunction with medications that affect CYP3A4 in other ways. In a phase 1, open-label, sequential study in healthy volunteers, rifampicin was used to examine the impact of CYP3A4 induction on lenvatinib pharmacokinetics [22]. After multiple doses of rifampicin, the Cmax estimates of free lenvatinib were 8.7% higher than that of lenvatinib alone, which fell below the prespecified lower boundary for bioequivalence. This contrasts with the effect of rifampicin on other tyrosine kinase inhibitors, which exhibit more profound decreases in Cmax and AUC. These results indicate that the clinical implications of coadministration of lenvatinib with CYP3A4 inducers are minimal. Similarly, examination of lenvatinib in combination with an inhibitor of CYP3A4 (ketoconazole) also revealed interactions that were not clinically significant [23]. However, as the previous studies assessed lenvatinib pharmacokinetic interactions in healthy patients, the current study in a clinically relevant population of patients with solid tumors is particularly valuable.
The goal of an interaction study such as this one is to provide clinical information related to increases or decreases in exposure to study drugs when coadministered with other therapeutics. This is crucial information in oncology as many cancer treatments are combination therapies. Lenvatinib in combination with everolimus is approved for the treatment of patients with advanced renal cell carcinoma following 1 prior antiangiogenic therapy [5]. Everolimus is a CYP3A4 substrate, and the FDA recommends caution or avoidance of concomitant administration with CYP3A4 inducers or inhibitors [24], so the potential for interaction is particularly important. In a phase 1 study assessing this combination treatment, no significant pharmacokinetic drug–drug interactions were observed [25]. In addition to their cancer therapies, many patients are taking concomitant medications to ameliorate other symptoms or to ease TEAEs. The information that lenvatinib does not have clinically significant effects on medications metabolized by CYP3A4 is critical for physicians in prescribing these additional therapies.
Selecting a study design for a drug–drug interaction study is complicated, with no single optimal design because of the complexities involved [26]. A crossover-study design is the preferred design of both the FDA and EMA [19, 20], but in studies with cancer patients who need to stay on the study drug, this design is unethical. Another potential design is a parallel-study design, but this is not recommended because of intersubject variability [19, 20]. As interaction studies in appropriate patient populations provide data that are more clinically relevant, a before/after design was selected for this study. This is the standard design for drug–drug interaction studies in patients and allowed the patients to keep receiving lenvatinib [27,28,29].
There are unique challenges and limitations involved in conducting drug–drug interaction studies in cancer patients. This study shares similar methodology with other cancer studies examining the impact of study drugs on midazolam pharmacokinetics. The effects of ridaforolimus, an mTOR inhibitor that exhibits anticancer activity, on midazolam activity were tested in an open-label, fixed-sequence study in 16 cancer patients [30]. Ridaforolimus was determined to have a minimal inhibitory impact on CYP3A, with no clinically significant implications [30]. Carfilzomib is an irreversible proteasome inhibitor that has demonstrated antitumor activity and its pharmacokinetic profile on midazolam was assessed in 18 patients with solid tumors, using a before/after study design over a 28-day cycle. No clinically significant effects of carfilzomib were seen on the pharmacokinetics of midazolam [31]. Oprozomib is a small molecule in development for multiple hematological malignancies and has shown activity as a potential CYP3A4 suppressor [32]. When studied in 18 patients with solid tumors, clinically relevant inhibition was not seen. Uniquely, this study utilized a design in which there was a washout period when patients received neither midazolam nor the study drug [32].
In addition to overall study design, performing these analyses on cancer patients instead of healthy controls poses other challenges, including the need to reduce burden on the patient. In this study, an effort was made to ease the demands made on patients by reducing the number of samples that would need to be taken, particularly overnight. For this reason, samples were taken frequently between 0 and 10 h after administration of midazolam, and then not again until 24 h, reducing the amount of available plasma concentration–time data. However, because the half-life of lenvatinib is 28 h and the tmax is 1–4 h after dosing [33], we believe this lengthy period between samples did not have a significant impact on the robustness of the pharmacokinetic data collected.
The number of patients in this study is aligned with the numbers in other drug–drug interaction studies [34], and a total of 18 patients was calculated as necessary to detect a 25% difference in the mean AUC between midazolam administered alone versus in combination with lenvatinib. In addition, to be classified as an inhibitor according to the FDA guidance, the differences in AUC are on a larger scale and therefore a lower-powered study is sufficient to detect changes. The FDA guidance states that a strong inhibitor increases the AUC by ≥ 5-fold, a moderate inhibitor increases the AUC by < 5 to ≥ 2-fold, and a weak inhibitor increases the AUC by < 2 to ≥ 1.25-fold [19]. If a drug displays an increase of < 1.25-fold on the AUC, it is not considered to be an inhibitor. As results from this study of the impact of lenvatinib on midazolam pharmacokinetics show an increase of 1.15-fold on the GMR of the AUC, it falls below the limit to be considered an inhibitor.
For this study, a noncompartmental analysis was chosen. This method is the preferred method for pharmacokinetic analysis when the goal is to obtain exposure levels following drug administration. This method has few underlying assumptions, and is appropriate for conditions when data are limited by a small number of patients or samples [21].
5 Conclusions
The results of this study demonstrate that coadministration of lenvatinib with midazolam appears to have no clinically relevant effects on the pharmacokinetics of midazolam, a CYP3A4 substrate, either following a single dose of lenvatinib or when lenvatinib concentrations were at steady state after multiple doses. The results of this study are consistent with physiologically based pharmacokinetic modeling, showing good correlation between the modeling and in vivo clinical data [18]. No new safety concerns were identified during this study, and lenvatinib was generally well tolerated. The most frequently reported TEAEs and treatment-related TEAEs (fatigue, diarrhea, and hypertension) were manageable and consistent with the known safety profile of lenvatinib.
References
Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res. 2014;2014:638747.
Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18.
Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14:5459–65.
Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–71.
Lenvima (lenvatinib) [prescribing information]. Woodcliff Lake, NJ, USA: Eisai Inc.; 2019.
Lenvima 4 mg hard capsules [summary of product characteristics]. Hertfordshire, UK: Eisai Europe Limited.
Yu R, Wang M, Zhu X, Sun Z, Jiang A, Yao H. Therapeutic effects of lenvatinib in combination with rAd-p53 for the treatment of non-small cell lung cancer. Oncol Lett. 2018;16:6573–81.
Suyama K, Iwase H. Lenvatinib: a promising molecular targeted agent for multiple cancers. Cancer Control. 2018;25:1073274818789361.
Sevrioukova IF, Poulos TL. Understanding the mechanism of cytochrome P450 3A4: recent advances and remaining problems. Dalton Trans. 2013;42:3116–26.
Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine. 2007. https://drug-interactions.medicine.iu.edu. Accessed 2 Dec 2019.
Midazolam Hydrochloride Syrup [prescribing information]. Columbus, OH, USA: Roxane Laboratories; 2012.
Midazolam Injection USP. Product Monograph. 2017. https://www.pfizer.ca/sites/g/files/g10050796/f/201712/2017.10.25_Midazolam_PM_WP_E_210318.pdf. Accessed 2 Aug 2019.
Wandel C, Böcker R, Böhrer H, Browne A, Rügheimer E, Martin E. Midazolam is metabolized by at least three different cytochrome P450 enzymes. Br J Anaesth. 1994;73:658–61.
Paulson SK, Wood-Horrall RN, Hoover R, Quintas M, Lawrence LE, Cammarata SK. The pharmacokinetics of the CYP3A substrate midazolam after steady-state dosing of delafloxacin. Clin Ther. 2017;39:1182–90.
Wen C, Zhuang Z, Song H, Tong S, Wang X, Lin Y, et al. Metabolism of liver CYP450 and ultrastructural changes after long-term administration of aspirin and ibuprofen. Biomed Pharmacother. 2018;108:208–15.
Garg V, Shen J, Li C, Agarwal S, Gebre A, Robertson S, et al. Pharmacokinetic and drug-drug interaction profiles of the combination of tezacaftor/Ivacaftor. Clin Transl Sci. 2019;12:267–75.
Xiao JJ, Nowak D, Ramlau R, Tomaszewska-Kiecana M, Wysocki PJ, Isaacson J, et al. Evaluation of drug-drug interactions of rucaparib and CYP1A2, CYP2C9, CYP2C19, CYP3A, and P-gp substrates in patients with an advanced solid tumor. Clin Transl Sci. 2019;12:58–655.
Schuck E, Yu Y, Lai G, Aluri J, Shumaker R, Reyderman L, et al. Physiologically based pharmacokinetic modeling of lenvatinib correctly predicts lack of significant CYP3A-mediated drug–drug interaction with midazolam. In: Poster presented at the Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, March 21–24, 2018, Orlando, FL, USA.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications Guidance for Industry. 2017. https://www.fda.gov/media/82734/download. Accessed 2 Aug 2019.
Committee for Human Medicinal Products, European Medicine Agency. Guideline on the Investigation of Drug Interactions. 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Accessed 2 Aug 2019.
Gabrielsson J, Weiner D. Non-compartmental analysis. Methods Mol Biol. 2012;929:377–89.
Shumaker R, Aluri J, Fan J, Martinez G, Thompson GA, Ren M. Effect of rifampicin in the pharmacokinetics of lenvatinib in health adults. Clin Drug Investig. 2014;34:651–9.
Shumaker R, Aluri J, Fran J, Martinez G, Thompson GA, Ren M. Effects of ketoconazole on the pharmacokinetics of lenvatinib (E7080) in healthy participants. Clin Pharmacol Drug Dev. 2015;4:155–60.
Afinitor (everolimus) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015.
Matsubara N, Naito Y, Nakano K, Fujiwara Y, Ikezawa H, Yusa W, et al. Lenvatinib in combination with everolimus in patients with advanced or metastatic renal cell carcinoma: a phase 1 study. Int J Urol. 2018;25:922–8.
Lewis LD. Drug-drug interactions: is there an optimal way to study them? Br J Clin Pharmacol. 2010;70:781–3.
Gupta N, Hanley MJ, Venkatakrishnan K, Bessudo A, Rasco DW, Sharma S, et al. Effects of strong CYP3A inhibition and induction on the pharmacokinetics of ixazomib, an oral proteasome inhibitor: results of drug-drug interaction studies in patients with advanced solid tumors or lymphoma and a physiologically based pharmacokinetic analysis. J Clin Pharmacol. 2018;58:180–92.
Han TH, Gopal AK, Ramchandren R, Goy A, Chen R, Matous JV, et al. CYP3A-mediated drug-drug interaction potential and excretion of brentuximab vedotin, an antibody-drug conjugate, in patients with CD30-positive hematologic malignancies. J Clin Pharmacol. 2013;53:866–77.
Chow LQ, Smith DC, Tan AR, Denlinger CS, Wang D, Shepard DR, et al. Lack of pharmacokinetic drug-drug interaction between ramucirumab and paclitaxel in a phase II study of patients with advanced malignant solid tumors. Cancer Chemother Pharmacol. 2016;78:433–41.
Stroh M, Talaty J, Sandhu P, McCrea J, Patnaik A, Tolcher A, et al. Lack of meaningful effect of ridaforolimus on the pharmacokinetics of midazolam in cancer patients: model prediction and clinical confirmation. J Clin Pharmacol. 2014;54:1256–62.
Wang Z, Yang J, Kirk C, Fang Y, Alsina M, Badros A, et al. Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab Dispos. 2013;41:230–7.
Ou Y, Xu Y, Gore L, Harvey RD, Mita A, Papadopoulos KP, et al. Physiologically-based pharmacokinetic modelling to predict oprozomib CYP3A drug-drug interaction potential in patients with advanced malignancies. Br J Clin Pharmacol. 2019;85:530–9.
Hussein Z, Mizuo H, Hayato S, Namiki M, Shumaker R. Clinical pharmacokinetic and pharmacodynamic profile of lenvatinib, an orally active, small-molecule, multitargeted tyrosine kinase inhibitor. Eur J Drug Metab Pharmacokinet. 2017;42:903–14.
Rekić D, Reynolds KS, Zhao P, Zhang L, Yoshida K, Sachar M, et al. Clinical drug-drug interaction evaluations to inform drug use and enable drug access. J Pharm Sci. 2017;106:2214–8.
Acknowledgements
This study was funded by Eisai Inc., and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. The authors thank Di Li, PhD; Javier Ferrada, MS; and Nicholas Batty, MD, for their contributions to this study. Medical writing assistance was provided by Caroline Leitschuh, PhD, of Oxford PharmaGenesis Inc., Newtown, PA, USA with funding provided by Eisai Inc., and Merck Sharp & Dohme Corp.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by Eisai Inc., Woodcliff Lake, NJ, USA and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Medical writing assistance was provided by Caroline Leitschuh, PhD, of Oxford PharmaGenesis Inc., Newtown, PA, USA and funded by Eisai Inc., and Merck Sharp & Dohme Corp.
Conflict of interest
Robert Shumaker is a former employee of Eisai Inc., Woodcliff Lake, NJ, USA. Min Ren, Jagadeesh Aluri, Corina Dutcus, Christian Rance and Cixin He are currently employed by Eisai Inc., Woodcliff Lake, NJ, USA.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the International Conference on Harmonisation of Pharmaceuticals for Human Use, the United States Code of Federal Regulations, the Institutional Review Boards of the University of Pennsylvania (FWA 00004028) and the University of Texas, San Antonio (FWA 00005928), the Western Institutional Review Board, and the Biomedical Research Alliance of New York, LLC (FWA00003433), and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shumaker, R., Ren, M., Aluri, J. et al. An Open-Label Phase 1 Study to Determine the Effect of Lenvatinib on the Pharmacokinetics of Midazolam, a CYP3A4 Substrate, in Patients with Advanced Solid Tumors. Eur J Drug Metab Pharmacokinet 45, 373–383 (2020). https://doi.org/10.1007/s13318-020-00607-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-020-00607-7