
Abstract
Objectives
The construction and validation of a set of Yarrowia lipolytica CRISPR/Cas9 vectors containing six different markers that allows virtually any genetic background to be edited, including those of wild-type strains.
Results
Using the Golden Gate method, we assembled a set of six CRISPR/Cas9 vectors, each containing a different selection marker, to be used for editing the genome of the industrial yeast Y. lipolytica. This vector set is available via Addgene. Any guide RNA (gRNA) sequence can be easily and rapidly introduced in any of these vectors using Golden Gate assembly. We successfully edited six different genes in a variety of genetic backgrounds, including those of wild-type strains, with five of the six vectors. Use of these vectors strongly improved homologous recombination and cassette integration at a specific locus.
Conclusions
We have created a versatile and modular set of CRISPR/Cas9 vectors that will allow any Y. lipolytica strain to be rapidly edited; this tool will facilitate experimentation with any prototroph wild-type strains displaying interesting features.
Similar content being viewed by others


Introduction
Yarrowia lipolytica is widely used as a microbial cell factory chassis in the development of industrial applications aiming to produce fatty acids, organic acids, or enzymes (Nicaud 2012; Madzak 2015; Ledesma-Amaro and Nicaud 2016). Although many engineering tools are now available (Larroude et al. 2018), the high rate of non-homologous end joining (NHEJ) impairs the efficiency of targeted genome modification. Moreover, limitations related to selection markers and the need to recycle them make engineering efforts even more time-consuming, and it can be challenging to generate highly modified strains with traits that correspond to the demands of industrial processes. CRISPR/Cas9 technology is being continually refined, and it was rapidly implemented in Saccharomyces cerevisiae (DiCarlo et al. 2013). It has also been successfully used in Y. lipolytica, with the aim of overcoming the aforementioned limitations and accelerating engineering cycles.
Several CRISPR/Cas9 systems dedicated to Y. lipolytica have recently been described. Both an RNA polymerase II (Pol II) transcription system and RNA polymerase III (Pol III) elements have been set up. Gao et al. (Gao et al. 2016) developed a Pol II gene disruption system that has successfully yielded single to triple mutations in a single step. Schwartz et al. (Schwartz et al. 2016) used a Pol III system and developed an efficient synthetic Pol III-tRNA hybrid promoter for gene disruption. In addition, a guide RNA (gRNA) system involving the expression of orthogonal T7 polymerase was exported to Y. lipolytica; it was based on the pCRISPRyl vector of Schwartz et al. (Morse et al. 2018). Structure–function relationships study and methodological research focused on single point mutations has also been described (Borsenberger et al. 2018). In addition, alternative systems, such as using defective Cas9 (CRISPRi) to inhibit expression, CRISPRa to activate gene expression, and the dual CRISPR/Cas9 system to excise and integrate genes have been shown to function in Y. lipolytica (Schwartz et al. 2017a, 2018; Gao et al. 2018).
It is now well established that the CRISPR/Cas9 system functions properly in Y. lipolytica, and modular and robust tools are needed for this system to become the standard for editing the genome of this yeast species. Such tools are particularly lacking when it comes to engineering the diversity of wild-type strains. These strains are never used as there are no markers available for them, which hinders metabolic engineering. To date, researchers have mainly concentrated on “laboratory” strains for which auxotrophic markers are already available. However, it is now well known that wild-type isolates display a wide range of traits when it comes to producing lipids, citric acids, or polyols, for example (Egermeier et al. 2017; Quarterman et al. 2017; Carsanba et al. 2019); in these experiments, some of the isolates strongly outperformed the standard laboratory strains. Wild-type strains may also respond very differently to environmental conditions and control parameters (Egermeier et al. 2017).
Having selectable markers is crucial to the success of genome editing technologies. In Y. lipolytica, such markers were initially based on leucine (LEU2), uracil (URA3), lysine (LYS5) and adenine (ADE2) auxotrophies (Barth and Gaillardin 1996). The first dominant markers to be developed relied on the expression of the E. coli hph gene, which confers antibiotic resistance to hygromycin B (Barth and Gaillardin 1996). Additional markers have been developed more recently. They are based on the Streptomyces noursei Nat1 gene (which provides resistance to nourseothricin) (Kretzschmar et al. 2013); the Y. lipolytica AHAS gene (which provides resistance to the herbicide chlorimuron ethyl); the E. coli guaB gene (which provides resistance to mycophenolic acid) (Wagner et al. 2018); the Streptoalloteichus hindustanus ble gene (which provides resistance to zeocin) (Tsakraklides et al. 2018); and the phosphite dehydrogenase ptxD gene from Pseudomonas stutzeri (which allows Y. lipolytica to grow on potassium phosphite in phosphate-deficient media) (Shaw et al. 2016). Additional markers have been developed that are related to the ability to catabolize carbon sources (hereafter referred to as “catabolic selectable markers” [CSM]). These markers have the advantage of not being involved in essential metabolic pathways. For instance, the S. cerevisiae SUC2 gene, which encodes invertase, has been used to select transformants on sucrose media (Nicaud et al. 1989). More recently, a novel CSM was developed that is centered on the EYK1 gene, which encodes an erythrulose kinase (Vandermies et al. 2017). This enzyme participates in an early step in erythritol catabolism and is essential for cell growth on erythritol-based medium (Carly et al. 2017).
Here, we describe the construction and validation of a set of seven CRISPR/Cas9 vectors, which each contain a different selection marker. They allow genome editing within virtually any genetic background and are particularly useful for working with wild-type strains, thanks to the panel of dominant markers they represent. Having access to such tools is especially important when it is necessary to integrate multiple cassettes to generate large pathways, which involves the use of multi-auxotrophic strains. gRNA sequences can easily be cloned into these vectors via the Golden Gate method. Consequently, these replication-based CRISPR/Cas9 vectors can be employed to knock out auxotrophic or CSM genes and thus increase the panel of markers available in any strain.
Materials and methods
Strains and media
The Escherichia coli and Y. lipolytica strains and plasmids used in this study are listed in Table S1. E. coli strain DH5α was used for cloning and plasmid propagation. The transformation of chemically competent E. coli cells was performed using a heat shock protocol. Cells were grown at 37 °C with constant shaking on 5 ml of LB medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) that contained ampicillin (100 μg/ml) or kanamycin (50 μg/ml) for plasmid selection. For yeast selection and growth, minimal YNBD medium containing 10 g/L glucose (Sigma), 1.7 g/L yeast nitrogen base (YNBww; Difco), 5.0 g/L NH4Cl, and 50 mM phosphate buffer (pH6.8) was used. To meet auxotrophic requirements, uracil (0.1 g/L), lysine (0.8 g/L), and leucine (0.1 g/L) were added to the culture medium when necessary. Erythritol and lysine were used as carbon sources at concentrations of 10 g/L and 0.1 g/L, respectively, to allow the selection of Δeyk1 and Δeyd1 mutants; lysine was used as an additional carbon source to boost growth in the absence of glucose, but in a concentration not sufficient for Δeyk1 and Δeyd1 to grow in presence of erythritol and absence of glucose. An oleic acid emulsion (0.05%) was used as a carbon source for Δmfe selection. Tributyrin YNB medium was utilized for Δlip2 selection as described in (Pignede et al. 2000). For antibiotic selection, hygromycin (250 μg/ml) or nourseothricin (400 μg/ml) was added to rich YPD medium (20 g/L Bacto Peptone, 10 g/L yeast extract, and 20 g/L glucose). Solid media were prepared by adding 15 g/L agar (Invitrogen) to liquid media.
Construction of acceptor vectors for gRNA cloning
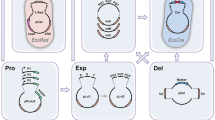
Six fragments were used to assemble the CRISPR/Cas9 acceptor vectors. Fragment 1 was the pSB1A3 bacterial plasmid containing the ampicillin resistance gene and the ColE1 region for selection and propagation in E. coli. Fragment 2 was the sgRNA module with the BsmBI recognition sites flanking the red fluorescent protein (RFP) gene, which was used as a chromophore in E. coli and which replaced the 20-nt target sequence (Fig. 1b). Fragment 3 was the excisable marker flanked by the I-sceI sites and the LoxP/LoxR. Fragment 4 was a 227-bp portion of the ARS68 sequence described in (Fournier et al. 1991), which allows plasmid replication and segregation in Y. lipolytica. Another 105-bp portion of the ARS68 sequence was present at the 3′ end of the sgRNA module and was thus cloned into the vector as part of fragment 2. Fragment 5 was the strong hybrid promoter p8UAS1TEF described by (Celinska et al. 2017), which is used in the expression of the endonuclease. Fragment 6 was the codon-optimized Streptococcus pyogenes Cas9-SV40 system including the CYC-t terminator described by Schwartz et al. (Schwartz et al. 2016). The internal BsmBI and BsaI sites were removed from all the fragments, which were then synthetized by Twist Bioscience or GeneMill.

Schematic draw showing an example of a CRISPR/Cas9 acceptor vector assembled via the Golden Gate method. a Assembly of the six fragments: the E. coli bacterial component (fragment 1); the sgRNA region containing the E. coli red fluorescent protein chromophore (RFP) gene flanked by the BsmBI sites (fragment 2); the excisable Y. lipolytica marker ylLEU2ex (fragment 3); the Y. lipolytica centromeric region ylARS1 (fragment 4); the promoter region P2_8UAS TEF (fragment 5); and the Cas9-SV40 region (fragment 6). b Cutting with BsmBI releases the RFP gene and permits the assembly of the 20-nt target sequence at that same location. c BsaI and BsmBI overhang sequences
To build fragment 2, the fragment carrying the RFP gene was amplified using primer pair RFP-SfiI-Fw/RFP-SfiI-Rv, and the plasmid GGE029 was employed as a template (Supplementary Table S1). Then, the RFP gene flanked by the SfiI sites was inserted into the sgRNA module (JME4315), which itself included the same SfiI site, giving rise to sgRNA::RFP JME4366 (Table 1).
The acceptor vectors were assembled using the Golden Gate method in accordance with the Larroude et al. protocol (Larroude et al. 2019). Plasmid DNA was extracted using a commercial miniprep kit (NucleoSpin® Plasmid, Macherey–Nagel) in accordance with the manufacturer’s instructions. Correct assembly was verified by colony PCR and by plasmid digestion with the BglII restriction enzyme. To build the different acceptor vectors, alternative versions of fragment 3 with the different Y. lipolytica markers were employed (from 3a to 3f, see Table 1).
Digestion of the acceptor vectors with BsmBI released the RFP gene and allowed the 20-nt target sequence to be inserted at that position. This process made it possible to rapidly visually screen for positive E. coli clones that had correctly assembled the CRISPR/Cas9 vectors, in which the gRNA had replaced the RFP gene.
gRNA design and plasmid construction
The gRNA was designed with the CRISPOR tool (https://crispor.tefor.net/). We chose target sequences with high efficiency scores and low numbers of predicted off-target sites; our preference was for target sequences in the middle of the open reading frame. The target sequences were 20 nt in length and did not include the PAM sequence. The gRNA was introduced into the acceptor vectors by annealing two overlapping oligonucleotides that generated overhangs matching those of the BsmBI site of the acceptor vectors. The oligonucleotides had the following structure: forward oligonucleotide: 5′TTCGATTCCGGGTCGGCGCAGGTTGNNNNNNNNNNNNNNNNNNNNGTTTTA 3′ reverse oligonucleotide: 5′GCTCTAAAACNNNNNNNNNNNNNNNNNNNNCAACCTGCGCCGACCCGGAAT 3′, where N represents the 20-nt target sequence. The underlined letters indicate the 4-nt overhang. The complementary gRNA oligonucleotides were phosphorylated and annealed as follows: we mixed 1 μL T4 Kinase (New England Biolabs, Ipswich, MA), 1 μL forward oligonucleotide (100 μM), 1 μL reverse oligonucleotide (100 μM), 1 μL T4 Ligase Buffer (10X; New England Biolabs), and 6 μL H2O; we incubated the mixture at 37 °C for 30 min and then heated the mixture so it stayed at 95 °C for 5 min; and we allowed the temperature to drop back down to 25 °C at a rate of 5 °C min−1. The reaction mixture was diluted with water (ratio of 1:200) before being assembled with the CRISPR acceptor vector.
The gRNA double-stranded insert was assembled with the pGGA_CRISPRyl acceptor vector via the Golden Gate method as follows: we mixed 2 μL gRNA insert, 100 ng pGGA_CRISPRyl, 2 μL T4 Ligase Buffer (New England Biolabs), 1 μL BsmBI (New England Biolabs), 1 μL T7 Ligase (New England Biolabs), and 20 µl H2O, and we incubated the mixture in a thermocycler using the following program: [5 min at 55 °C, 5 min at 16 °C] × 30, 5 min at 50 °C, and 5 min at 80 °C. We used 10 μL of the assembly mix to transform E. coli and then grew the bacteria on LB ampicillin plates. Plasmid DNA was extracted using a commercial miniprep kit (NucleoSpin® Plasmid, Macherey–Nagel) in accordance with the manufacturer’s instructions. Correct assembly of the positive transformants (white E. coli) was verified either by colony PCR or by plasmid digestion with the BglII restriction enzyme. The status of positive clones was confirmed via sequencing.
Transformation of the pGGA_CRISPRyl-gRNA plasmids to perform gene editing in Y. lipolytica
Transformation of Y. lipolytica was performed using the lithium-acetate method (Barth and Gaillardin 1996). Cells were either plated directly onto selective media for the direct selection of transformants (no outgrowth step), or an outgrowth step was performed on selective liquid media and allowed recovery. In the outgrowth step, cells were cultured in 5 mL of selective media for 24 h and then diluted to obtain 50–100 colonies per plate of rich YPD medium. The transformants could then be tested for the desired phenotype. The gene disruption success rate was determined by counting the number of colonies that showed the expected phenotype; the status of positive clones was confirmed via sequencing. After the screening step, the clones with the desired phenotype were grown in rich liquid YPD medium for 12–24 h to cure the CRISPR\Cas9 plasmid.
Transformation using the pGGA_CRISPRyl-gRNA plasmids and the deletion cassette for specific locus integration
The deletion cassette was composed of a URA3ex expression cassette flanked upstream and downstream by the homologous recombination sequences for the GSY1 gene. Overall structure of the cassette and construction has been described elsewhere (Fickers et al. 2003). Y. lipolytica was transformed using the lithium-acetate method (Barth and Gaillardin 1996). Then, 500 ng of the CRISPR-Cas9 vector was co-transformed with 500 ng of the NotI-digested deletion cassette. The transformation reaction was inoculated in 9 mL of selective liquid medium and cultured for 48 h. One ml of the culture was then transferred into YPD medium for 24 h to allow plasmid curing. Finally, the culture was diluted and plated so as to obtain 50–100 colonies per plate.
When the deletion cassette was transformed without the CRISPR\Cas9 vector, no outgrowth was performed, and 200 µL of the transformation reaction was directly grown on plates containing selective agar.
Mutant phenotype analysis
After the transformation with the CRISPR\Cas9 vector, the outgrowth step, and the plating, the colonies could undergo phenotype screening.
gsy1 disruption
When gsy1 had been successfully disrupted, the phenotype was an absence of glycogen accumulation, which could be visualized using staining with Lugol’s iodine (prepared by mixing water solutions of 2% KI and 1% I2 at a ratio of 1:1). Colonies in which gsy1 had been successfully disrupted remained clear, while colonies containing active GSY1 were brown. There are two screening options. The clones can be tested on their plates, by adding 4 mL of Lugol’s iodine. They can also be tested in 96-well microplates after 24 h of culture on YPD medium: 30 µL of Lugol’s iodine is added to each well after the supernatant culture has been eliminated.
eyk1 and eyd1 disruption
Transformants were grown on both YPD and YNB-erythritol-lysine media for 48 h at 28 °C. Then, we assessed whether clones were able to use erythritol as a carbon source. When a clone grew on YPD but not on YNB-erythritol-lysine, it indicated that eyk1 or eyd1 had been disrupted. The JMY7126 strain (Δeyk1) was used as the positive control.
mfe1 disruption
Clones in which mfe1 had been disrupted should be unable to use oleic acid as a carbon source. Transformants were grown on both YPD and YNB-oleic acid media for 48 h at 28 °C. When a clone grew on YPD medium but not on YNB-oleic acid medium, it indicated that mfe1 had been disrupted. The strain JMY1888 (Δmfe1) was used as a positive control.
ura3 disruption
Transformants were grown on both YPD medium and YNB medium without uracil for 48 h at 28 °C. When a clone grew on YPD but not on YNB without uracil, it indicated that ura3 had been disrupted.
lip2 disruption
The LIP2 gene encodes the main extracellular lipase, so Δlip2 strains display a reduced halo of triglyceride hydrolysis on YNB medium containing tributyrin (Pignede et al. 2000). Transformants were grown on YNB medium containing tributyrin for 48 h at 28 °C. Clones with strongly reduced halos were considered to carry a disrupted version of LIP2.
Colony PCR and sequence-based verification of gene disruption
For positive clones, we verified gene disruption via sequencing. We employed specific primers that spanned the gRNA target regions (supplementary Table S2). First, the regions were amplified by colony PCR. Single colonies were picked with a tip and transferred to 2µL water and cells were lysed with a 15 min heating cycle in the thermocycler. The cells lysed were directly used in the standard PCR reaction. The amplified fragments were then purified using gel extraction and PCR purification kits (NucleoSpin® Gel and PCR Clean-Up, Macherey–Nagel) and sequenced. The sequences were aligned against reference sequences to identify the mutations in the target sequence.
Results
Construction of a versatile CRISPR/Cas9 vector set
To extend the set of synthetic biology tools available for Y. lipolytica, we wished to develop a systematic backbone method for building a set of CRISPR/Cas9 vectors. Our design was based on the basal structure of the plasmid developed by Schwartz et al. (Schwartz et al. 2016), and the Golden Gate method made it possible to assemble and switch out different parts. We took advantage of a large set of Golden Gate bricks that were recently made available for Y. lipolytica (Larroude et al. 2019). For our specific CRISPR/Cas9 vector set, the necessary parts include i) a bacterial plasmid containing the ampicillin resistance gene and the ColE1 region for selection and propagation in E. coli; ii) a Y. lipolytica selection marker; iii) the ARS/CEN fragment allowing plasmid replication in Y. lipolytica; iv) a Y. lipolytica promoter for endonuclease expression; v) the Cas9 endonuclease optimized for Y. lipolytica; and vi) a region for cloning gRNA. In association with the latter, we implemented a direct screening method for verifying that the recombinant plasmid had incorporated the gRNA: we integrated a RFP chromophore gene at the gRNA cloning site that would be later released, allowing the assembly of the 20-nt target sequence at that same location (Celinska et al. 2017). The set of bricks designed and used to assemble the CRISPR/Cas9 acceptor vectors are listed in Table 1. These acceptor vectors were first put together using BsaI overhang sequences, as described in Larroude et al. (Larroude et al. 2019). These acceptor vectors were then ready to be assembled with the gRNA fragment of choice using the BsmBI overhang sequences, as described in the Material and Methods. The overall design is depicted in Fig. 1; the Golden Gate overhang sequences, the BsaI overhang sequences for backbone construction, and the BsmBI overhang sequences for gRNA cloning are indicated.
Using the pool of Golden Gate bricks, we built a set of acceptor vectors that were ready for immediate use in gRNA cloning employing different markers. We used LEU2ex, URA3ex, and LYS5ex for the auxotrophic markers; NATex and HPHex for the antibiotic markers; and EYK1ex for the CSM markers. All the acceptor vectors are listed in Table 2.
Validation of genome editing
We tested our CRISPR/Cas9 system by first targeting genes that can be used as markers: EYK1, EYD1, and URA3. The EYK1 gene encodes erythrulose kinase, which is involved in erythritol catabolism. The disruption of this gene allows the selection of strains that cannot use erythritol as their sole carbon source (Carly et al. 2017). The EYD1 gene encodes erythritol dehydrogenase, which is also involved in erythritol catabolism (Carly et al. 2018). The URA3 gene is involved in uracil metabolism. The disruption of EYK1 and EYD1 increased the strength of recently developed erythritol-inducible promoters pEYK1 and pEYD1 (Trassaert et al. 2017; Park et al. 2019). It is therefore particularly useful to disrupt these genes in strains that contain expression cassettes based on these promoters.
We then extended our testing to include genes related to lipid metabolism: MFE2, GSY1, and LIP2. The MFE2 gene encodes the multifunctional enzyme, which is involved in the fatty acid degradation pathway. Its inactivation results in cells that are unable to use fatty acids as their sole carbon source (Smith et al. 2000). The GSY1 gene encodes glycogen synthase, which is involved in glycogen synthesis. Its inactivation results in carbon storage being redirected from sugars (glycogen) to lipids (triacyl glycerol) (Bhutada et al. 2017). The LIP2 gene encodes lipase, which is involved in external lipid degradation (Pignede et al. 2000).
gRNAs were designed for all these targets and then assembled in the CRISPR/Cas9 vector JME4390. All gene editing was performed in the strain Po1d (JMY195). Table 3 shows the editing success rate (i.e., following phenotype screening and sequence-based verification; see the Materials and Methods section). Editing success was highly variable (7–70%), and it depended on the target gene. It was also dependent on the gRNA sequence (data not shown), a finding reported in a large scale study in Y. lipolytica (Schwartz et al. 2019). Because it was easy to screen and had a median editing success rate, we chose to target GSY1 using our CRISPR/Cas9 vector set. More specifically, the CRISPR/Cas9-gGSY1 vectors were used in different genetic backgrounds adapted to the six markers (Table 4). The results show that editing success was highly dependent on the marker/strain combination used and ranged from 4% (JMY195 with the HPHex marker) to 81% (JMY330 with the LEU2ex marker and JMY195 with the NATex marker). For one marker, EYK1ex, no positive clones were obtained (Table 4). For this strain/marker combination, either editing failed completely or the success rate was below 1% and thus undetectable.
In Y. lipolytica, homologous recombination is not very efficient. To confirm that our set of CRISPR/Cas9 vectors could help integrate DNA via homologous recombination, we compared the rate of homologous recombination with and without co-transformation by CRISPR/Cas9 vectors in a standard genetic background and in a Y. lipolytica strain deleted for ku70, which has shown improved homologous recombination (Verbeke et al. 2013). A classical disruption cassette composed of a URA3 expression cassette flanked by 1-kb homologous regions upstream and downstream from the GSY1 gene—was used to transform Po1d and Po1d Δku70. Table 5 shows that using a CRISPR/Cas9 vector in tandem with the integration cassette strongly improved homologous recombination. The success rate was nearly 100% in the Δku70 background and 83% in the Po1d background. When the cassette alone was used, the success rate was only around 15%. High success rates were obtained in a similar experiment using the same backbone plasmid (Schwartz et al. 2016). The low level of homologous recombination in Y. lipolytica is a drawback in standard deletion and targeted integration procedures. Moreover, the need for a long flanking region complicates cloning, but reducing the size of the homologous region strongly reduces the rate of homologous recombination. To determine if using a CRISPR/Cas9 vector in tandem with a shortened homologous region could improve the rate of correct integration, similar experiments were performed using homologous regions of different lengths (100 bp and 50 bp) located both upstream and downstream from the GSY1 gene. When the 100-bp region was used in the absence of the CRISPR/Cas9 vector, no positive clones resulted from the deletion of the GSY1 gene (Table 5). However, when the CRISPR/Cas9 vector was employed, the success rate was reasonable (25%); it was very high (88%) in the Δku70 background. When the 50-bp region was used, there were no positive clones (Table 5). In short, using a CRISPR/Cas9 vector makes it possible to strongly reduce the size of the homologous recombination region (down to 100 bp) and still obtain a reasonable rate of successful insertion with or without using a Δku70 background.
One of the advantages of having access to a large set of markers is that it becomes faster to carry out multiple deletions in a single strain. We therefore wished to verify that we could edit multiple genome locations by transforming yeast using two CRISPR/Cas9 plasmids in tandem. In the strain JMY5217 (Po1d Lys−Leu−), we simultaneously disrupted gsy1 and ura3 using the JME 4392 (CrCas9-Leu2-gGSY) JME 4453 (CrCas9-Lys5-gURA), respectively. For the 48 colonies tested, the success rate was 50% for ura3 and 85% for gsy1. All the ura3 knockouts were also gsy1 knockouts. When the markers were inversed, by using JME4425 (CrCas9-Lys5-gGSY) and JME 4452 (CrCas9-Leu2-gURA), our success rate was nil for ura3 and 62% for gsy1. These results show that, in Y. lipolytica, it is possible to carry out simultaneous transformation employing multiple CRISPR/Cas9 vectors that have the same backbone but that express different markers. However, some gRNA-marker combinations appear less efficient than others as already observed (Table 4). In particular, Leu2-gURA was not successful in our hand in the JMY5217 background while it is in the JMY195 background (Table 3).
Genome editing in wild-type strains using CRISPR/Cas9 vectors
Most of the genetic engineering that takes place in Y. lipolytica involves a small subset of laboratory strains that have been specifically developed for this purpose by introducing auxotrophies. Because standard engineering methods therefore rely on auxotrophies, limited use has been made of the diverse characteristics displayed by different Y. lipolytica wild-type strains. However, as mentioned in the introduction, wild-type strains can outperform laboratory strains. They are therefore a better chassis for carrying out further modifications. One of our objectives in developing this set of CRISPR/Cas9 vectors with dominant markers was to be able to perform genome editing in wild-type Y. lipolytica strains. We used our CRISPR/Cas9-hph vector, into which gsy1 gRNA had been introduced, to transform a collection of wild-type strains as a proof of principle. This plasmid was used to transform nine different wild-type strains representing a broad range of origins (see Supplementary Table S1). The rate at which we successfully disrupted gsy1 was determined using Lugol staining, as shown in Fig. 2 (A and B); the editing success rates are indicated in Table 6. Positive clones were sequenced to confirm editing success.

CRISPR/Cas9-mediated disruption of the GSY1 gene in wild-type strains. a Clones on plates before Lugol staining. b Clones on plates after Lugol staining. List of strains tested (from top left to bottom right): CBS 2074, CBS 6125, CLIB 791, CLIB 879, DBVPG 4400, DBVPG 5851, NCYC 3271, PYCC 4743 and IMUFRJ 50682
The editing success rate was determined based on the staining patterns of the transformants (dark vs. clear in response to Lugol staining). As variability in staining patterns was observed among strains (Fig. 2), we sequenced between one and three clear clones for each strain to validate the genome editing process. In all cases, we observed that genome editing occurred upstream from the PAM sequence, at the gRNA target sequence. In general, our CRISPR/Cas9 vectors with dominant markers were well suited to genome editing in wild-type strains. All the wild-type GSY1 genes were sequenced, and their sequences were compared to the CLIB122 sequence that was used to design the gRNA. No differences were seen in the gRNA target region, excluding potential mismatch bias. We observed dramatic differences in editing success: it ranged from 100% for the strains IMUFRJ 50682 and CBS 6125 to 0% for strains CLIB 791 and DBVPG 4400 (Table 6). This observation implies that it might be easier to genetically engineer some wild-type strains than others, but wild-type strains could still outperform some of the more widely used laboratory strains. We were able to easily transform all the wild-type strains with our vector even if two of them failed to show any signs of editing. All the strains tested are sensitive to hygromycin (growth inhibition between 60 and 80 µg/mL) and no major differences in sensitivity were observed that could explained differences in transformability and/or editing.
Discussion
Here, we describe how we used the Golden Gate method to assemble a set of CRISPR/Cas9 vectors, each containing a different marker, that can be employed to perform genome editing in Y. lipolytica. The vectors are now available via Addgene (129,656–129,661). In all these vectors, any gRNA sequence can be easily and rapidly introduced using Golden Gate assembly.
We built our system by taking advantage of the large set of bricks available for Y. lipolytica and a recently published Golden Gate protocol (Larroude et al. 2019). In our system, the Cas9 endonuclease could be placed under the control of different promoters that vary in strength and inducibility, which could be useful to temporally control expression. We decided to employ the 8UASpTEF promoter because our comparison of the results obtained with pTEF and 8UASpTEF found no significant differences in editing efficiency (data not shown). Borsenberger et al. showed that promoter strength was not critical and that pTEF provides adequate results compared to 8UASpTEF when it comes to the expression of the Cas9 endonuclease in Y. lipolytica (Borsenberger et al. 2018). The only constraint is that RNA structure must be compatible. Egermeier et al. (Egermeier et al. 2019) recently published a Golden Gate protocol for building Y. lipolytica CRISPR/Cas9 vectors, but its applicability is more limited than that of our protocol because it does not include a range of markers or a rapid-screening method (i.e., we used RFP as a negative reporter of assembly success). They were able to knockout LEU2 in a wild-type strain and showed that their system was functional (editing success rate: 6–25%). They used a different CRISPR/Cas9 system that is based on the HH ribozyme and a humanized Cas9 that is not codon optimized for Y. lipolytica (Gao et al. 2016). Systems utilizing HH ribozyme gRNA processing have been shown to be less efficient than systems utilizing the PolIII promoter system (Schwartz et al. 2016); we used the latter system here, and it was coupled with a Y. lipolytica optimized Cas9. Accordingly, in Y. lipolytica, codon-optimized Cas9 is expressed at higher levels than the humanized Cas9 when the polIII system is employed (Borsenberger et al. 2018).
We also used CRISPR/Cas9-mediated cutting to introduce an expression cassette at a specific locus. The editing success rate was much higher with CRISPR/Cas9 than without CRISPR/Cas9, and we were able to drastically reduce the length of the homologous recombination region without having to use a Δku background. This approach allowed us to avoid the pitfalls associated with the Δku background. It can also simplify and speed up metabolic engineering because integrating a large pathway at a specific locus can sometimes be a difficult task (Schwartz et al. 2017b), and using a marker can drastically improve efficiency. Marker-free integration, in which Cas9 cuts are repaired via homologous recombination, can work in Y. lipolytica, but the editing success rate is not consistent outside of the Δku background (Gao et al. 2016; Schwartz et al. 2017b; Holkenbrink et al. 2018).
All the vectors that we built were tested using different targets, and genome editing was successful in all cases but one (the EYK1ex marker). We took advantage of antibiotic markers to exploit the natural diversity of Y. lipolytica and validated our CRISPR/Cas9 vectors in a large number of wild-type strains. The editing success rate for gsy1 disruption differed dramatically among strains even though they harboured an identical gsy1 sequence. The colony morphology are diverse (Fig. 2) and may reflect physiology differences between strains. This can affect the rate of transformants if, for example, cell wall structures are different. Transformation procedure is the one setup and optimized for the laboratory strains W29 or CLIB122. Physiology differences may require different optimization for the other wild-type strains, which could improve transformation efficiency and ultimately editing efficiency. The phenotype screening also highlighted differences in staining patterns that probably reflect differences in glycogen accumulation. In Y. lipolytica, glycogen storage is detrimental to lipid accumulation (Bhutada et al. 2017), so these results indicate that gsy1 disruption (dark-stained strains) may have a great impact on lipid production. Another, less likely, hypothesis is that the penetration of Lugol’s iodine varied among strains.
To our knowledge, our set of CRISPR/Cas9 vectors is the most extensive to date for carrying out genome editing in Y. lipolytica.
References
Barth G, Gaillardin C (1996) The dimorphic fungus Yarrowia lipolytica. In: Wolf K (ed) Genetics, biochemistry and molecular biology of non conventional yeasts. Springer Verlag, Heidelberg, pp 313–388
Bhutada G, Kavšcek M, Ledesma Amaro R, Thomas S, Rechberger GN, Nicaud J-M, Natter K (2017) Sugar versus fat: elimination of glycogen storage improves lipid accumulation in Yarrowia lipolytica. FEMS Yeast Res 17(3):fox020. https://doi.org/10.1093/femsyr/fox020
Borsenberger V, Onésime D, Lestrade D, Rigouin C, Neuvéglise C, Daboussi F, Bordes F (2018) Multiple parameters drive the efficiency of CRISPR/Cas9-induced gene modifications in Yarrowia lipolytica. J Mol Biol 430:4293–4306. https://doi.org/10.1016/j.jmb.2018.08.024
Carly F, Gamboa Melendez H, Vandermies M, Damblon C, Nicaud J-M, Fickers P (2017) Identification and characterization of EYK1, a key gene for erythritol catabolism in Yarrowia lipolytica. Appl Microbiol Biotechnol 101:6573–6596. https://doi.org/10.1007/s00253-017-8412-4
Carly F, Steels S, Telek S, Vandermies M, Nicaud J-M, Fickers P (2018) Identification and characterization of EYD1, encoding an erythritol dehydrogenase in Yarrowia lipolytica and its application to bioconvert erythritol into erythrulose. Bioresour Technol 247:963–969. https://doi.org/10.1016/j.biortech.2017.09.168
Carsanba E, Papanikolaou S, Fickers P, Erten H (2019) Screening various Yarrowia lipolytica strains for citric acid production. Yeast 36:319–327. https://doi.org/10.1002/yea.3389
Celinska E, Ledesma-Amaro R, Larroude M, Rossignol T, Pauthenier C, Nicaud JM (2017) Golden gate assembly system dedicated to complex pathway manipulation in Yarrowia lipolytica. Microb Biotechnol 10:450–455. https://doi.org/10.1111/1751-7915.12605
DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM (2013) Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. https://doi.org/10.1093/nar/gkt135
Egermeier M, Russmayer H, Sauer M, Marx H (2017) Metabolic flexibility of Yarrowia lipolytica growing on glycerol. Front Microbiol 8:49. https://doi.org/10.3389/fmicb.2017.00049
Egermeier M, Sauer M, Marx H (2019) Golden gate based metabolic engineering strategy for wild-type strains of Yarrowia lipolytica. FEMS Microbiol Lett 366(4):fnz022. https://doi.org/10.1093/femsle/fnz022
Fickers P, Le Dall MT, Gaillardin C, Thonart P, Nicaud JM (2003) New disruption cassettes for rapid gene disruption and marker rescue in the yeast Yarrowia lipolytica. J Microbiol Methods 55:727–737
Fournier P, Guyaneux L, Chasles M, Gaillardin C (1991) Scarcity of ars sequences isolated in a morphogenesis mutant of the yeast Yarrowia lipolytica. Yeast 7:25–36. https://doi.org/10.1002/yea.320070104
Gao S et al (2016) Multiplex gene editing of the Yarrowia lipolytica genome using the CRISPR-Cas9 system. J Ind Microbiol Biotechnol 43:1085–1093. https://doi.org/10.1007/s10295-016-1789-8
Gao D, Smith S, Spagnuolo M, Rodriguez G, Blenner M (2018) Dual CRISPR-Cas9 cleavage mediated gene excision and targeted integration in Yarrowia lipolytica. Biotechnol J 13:e1700590. https://doi.org/10.1002/biot.201700590
Holkenbrink C, Dam MI, Kildegaard KR, Beder J, Dahlin J, Belda DD, Borodina I (2018) EasyCloneYALI: CRISPR/Cas9-based synthetic toolbox for engineering of the yeast Yarrowia lipolytica. Biotechnol J 13:e1700543. https://doi.org/10.1002/biot.201700543
Kretzschmar A, Otto C, Holz M, Werner S, Hubner L, Barth G (2013) Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr Genet 59:63–72. https://doi.org/10.1007/s00294-013-0389-7
Larroude M, Rossignol T, Nicaud JM, Ledesma-Amaro R (2018) Synthetic biology tools for engineering Yarrowia lipolytica. Biotechnol Adv 36:2150–2164. https://doi.org/10.1016/j.biotechadv.2018.10.004
Larroude M, Park Y-K, Soudier P, Kubiak M, Nicaud J-M, Rossignol T (2019) A modular Golden Gate toolkit for Yarrowia lipolytica synthetic biology. Microb Biotechnol 12:1249–1259. https://doi.org/10.1111/1751-7915.13427
Ledesma-Amaro R, Nicaud J-M (2016) Yarrowia lipolytica as a biotechnological chassis to produce usual and unusual fatty acids. Prog Lipid Res 61:40–50. https://doi.org/10.1016/j.plipres.2015.12.001
Madzak C (2015) Yarrowia lipolytica: recent achievements in heterologous protein expression and pathway engineering. Appl Microbiol Biotechnol 99:4559–4577. https://doi.org/10.1007/s00253-015-6624-z
Morse NJ, Wagner JM, Reed KB, Gopal MR, Lauffer LH, Alper HS (2018) T7 polymerase expression of guide RNAs in vivo allows exportable CRISPR-Cas9 editing in multiple yeast hosts. ACS Synthetic Biology 7:1075–1084. https://doi.org/10.1021/acssynbio.7b00461
Nicaud JM (2012) Yarrowia lipolytica. Yeast 29:409–418. https://doi.org/10.1002/yea.2921
Nicaud JM, Fabre E, Gaillardin C (1989) Expression of invertase activity in Yarrowia lipolytica and its use as a selective marker. Curr Genet 16:253–260
Park Y-K et al (2019) Engineering the architecture of erythritol-inducible promoters for regulated and enhanced gene expression in Yarrowia lipolytica. FEMS Yeast Res 19(1):foy105. https://doi.org/10.1093/femsyr/foy105
Pignede G, Wang H-J, Fudalej F, Seman M, Gaillardin C, Nicaud J-M (2000) Autocloning and amplification of LIP2 in Yarrowia lipolytica. Appl Environ Microbiol 66:3283–3289. https://doi.org/10.1128/aem.66.8.3283-3289.2000
Quarterman J, Slininger PJ, Kurtzman CP, Thompson SR, Dien BS (2017) A survey of yeast from the Yarrowia clade for lipid production in dilute acid pretreated lignocellulosic biomass hydrolysate. Appl Microbiol Biotechnol 101:3319–3334. https://doi.org/10.1007/s00253-016-8062-y
Schwartz CM, Hussain MS, Blenner M, Wheeldon I (2016) Synthetic RNA polymerase III promoters facilitate high-efficiency CRISPR–Cas9-mediated genome editing in Yarrowia lipolytica. ACS Synthetic Biology 5:356–359. https://doi.org/10.1021/acssynbio.5b00162
Schwartz C, Frogue K, Ramesh A, Misa J, Wheeldon I (2017a) CRISPRi repression of nonhomologous end-joining for enhanced genome engineering via homologous recombination in Yarrowia lipolytica. Biotechnol Bioeng 114:2896–2906. https://doi.org/10.1002/bit.26404
Schwartz C, Shabbir-Hussain M, Frogue K, Blenner M, Wheeldon I (2017b) Standardized markerless gene integration for pathway engineering in Yarrowia lipolytica. ACS Synthetic Biology 6:402–409. https://doi.org/10.1021/acssynbio.6b00285
Schwartz C, Curtis N, Lobs AK, Wheeldon I (2018) Multiplexed CRISPR activation of cryptic sugar metabolism enables Yarrowia lipolytica growth on cellobiose. Biotechnol J 13:e1700584. https://doi.org/10.1002/biot.201700584
Schwartz C et al (2019) Validating genome-wide CRISPR-Cas9 function improves screening in the oleaginous yeast Yarrowia lipolytica. Metab Eng 55:102–110. https://doi.org/10.1016/j.ymben.2019.06.007
Shaw AJ et al (2016) Metabolic engineering of microbial competitive advantage for industrial fermentation processes. Science 353:583–586. https://doi.org/10.1126/science.aaf6159
Smith JJ, Brown TW, Eitzen GA, Rachubinski RA (2000) Regulation of peroxisome size and number by fatty acid β-oxidation in the yeast Yarrowia lipolytica. J Biol Chem 275:20168–20178. https://doi.org/10.1074/jbc.M909285199
Trassaert M, Vandermies M, Carly F, Denies O, Thomas S, Fickers P, Nicaud J-M (2017) New inducible promoter for gene expression and synthetic biology in Yarrowia lipolytica. Microb Cell Fact 16:141. https://doi.org/10.1186/s12934-017-0755-0
Tsakraklides V et al (2018) High-oleate yeast oil without polyunsaturated fatty acids. Biotechnol Biofuels 11:131. https://doi.org/10.1186/s13068-018-1131-y
Vandermies M, Denies O, Nicaud J-M, Fickers P (2017) EYK1 encoding erythrulose kinase as a catabolic selectable marker for genome editing in the non-conventional yeast Yarrowia lipolytica. J Microbiol Methods 139:161–164. https://doi.org/10.1016/j.mimet.2017.05.012
Verbeke J, Beopoulos A, Nicaud J-M (2013) Efficient homologous recombination with short length flanking fragments in Ku70 deficient Yarrowia lipolytica strains. Biotechnol Lett 35:571–576. https://doi.org/10.1007/s10529-012-1107-0
Wagner JM, Williams EV, Alper HS (2018) Developing a piggyBac Transposon system and compatible selection markers for insertional mutagenesis and genome engineering in Yarrowia lipolytica. Biotechnol J 13:e1800022. https://doi.org/10.1002/biot.201800022
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 720824. We would like to thank Pr Maria Alice Zarur Coelho for providing the wild-type strain IMUFRJ50682 and Cécile Neuvéglise for providing the wild-type strains CBS 2074, CBS 6125, CLIB 791, CLIB 879, DBVPG 4400, DBVPG 5851, NCYC 3271, PYCC 4743.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Larroude, M., Trabelsi, H., Nicaud, JM. et al. A set of Yarrowia lipolytica CRISPR/Cas9 vectors for exploiting wild-type strain diversity. Biotechnol Lett 42, 773–785 (2020). https://doi.org/10.1007/s10529-020-02805-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02805-4