Dynamical Mean Field Studies of Infinite Layer Nickelates: Physics Results and Methodological Implications
 Hanghui Chen
Hanghui Chen Alexander Hampel3
Alexander Hampel3 - 1NYU-ECNU Institute of Physics, NYU Shanghai, Shanghai, China
- 2Department of Physics, New York University, New York, NY, United States
- 3Center for Computational Quantum Physics, Flatiron Institute, New York, NY, United States
- 4Department of Applied Physics and Applied Mathematics, Columbia University, New York, NY, United States
- 5European XFEL, Schenefeld, Germany
- 6Department of Physics, Columbia University, New York, NY, United States
This article summarizes recent work on the many-body (beyond density functional theory) electronic structure of layered rare-earth nickelates, both in the context of the materials themselves and in comparison to the high-temperature superconducting (high-Tc) layered copper-oxide compounds. It aims to outline the current state of our understanding of layered nickelates and to show how the analysis of these fascinating materials can shed light on fundamental questions in modern electronic structure theory. A prime focus is determining how the interacting physics defined over a wide energy range can be estimated and “downfolded” into a low energy theory that would describe the relevant degrees of freedom on the ∼
1 Introduction
The identification of a new class of superconductors is a momentous event. Ways in which the new superconductors are similar to or different from previously known materials can drive new insights into the microscopic origin of this fundamentally mysterious quantum many-body phenomenon. The discovery [1, 2] of superconductivity in layered copper-oxide materials sparked a revolution in condensed matter physics and materials science, because the transition temperatures were very high relative to other materials known at the time. Additionally, many aspects both of the superconductivity and of the non-superconducting (“normal state”) physics differed sharply from the predictions of conventional theory [3] in ways that made it obvious that interacting electron physics beyond mean field theory could have consequences of fundamental physics interest that approach (and in a few niche cases reach) commercial viability.
The very recent discovery [4] of superconductivity in the layered d9 nickelates was also momentous because the superconductivity was theoretically anticipated [5] on the basis of a chemical and structural similarity to the cuprates. Understanding the properties of the superconducting nickelates provides an immense scientific opportunity to sharpen our understanding of the relation between crystal structure and local chemistry on the one hand and important phenomena such as superconductivity on the other.
The relationship between physical phenomena and crystal structure/local chemistry is the central question in the theory of quantum materials. The theory of quantum materials is one instance of the quantum many-body problem, one of the grand scientific challenges of our time. The quantum many-body problem is in essence a problem of data compression: as is well known, many-particle quantum mechanics is formulated in a Hilbert space of a size that grows exponentially with the number of degrees of freedom while quantum entanglement in general and Fermi statistics in particular implies that delicate phase relationships between different states are of crucial importance. Incorporating the chemical realism needed to understand and predict material properties requires acting on this Hilbert space with a Hamiltonian matrix that involves a number of parameters which grows as a very high power of the number of degrees of freedom. All solutions of the quantum many-body problem require both reducing the number of degrees of freedom and the number of interaction parameters to a manageable level (“downfolding” the physics to a simpler systems, typically a “low energy theory”) and then handling the “model system” analysis of the still exponentially large and severely entangled Hilbert space of the downfolded theory.
Three decades of intense work have led to a rough consensus, generally although not universally accepted, that the low energy theory for the copper-oxide materials is some variant of the two dimensional one band Hubbard model. The relation of the model parameters to the precise chemistry and structure is reasonably well understood, and the properties of the simplest instantiations of this model are starting to come into focus [6–8]. The recent discovery of superconductivity in layered d9 nickelates such as NdNiO2 takes these questions to a new level. While many aspects of the crystal structure and chemistry are similar to those of the cuprates, in the nickelate family of materials multiple bands cross the Fermi surface and more interactions may be relevant. A crucial question is whether these effects are minor, so that the important physics of the nickelates is similar to that of the cuprates, or whether the low energy physics is richer and more complex in nickelates than in cuprates.
Various interacting models have been proposed for NdNiO2. The simplest interacting lattice model is a single Ni
Which of the plethora of theoretical models is most relevant is an important question. This review will present a perspective on what has been learned from the density functional theory (DFT) plus dynamical mean field theory (DMFT) about the many body electronic structure and the relevant low energy model and also what has been learned about the methods in the context of applying them to the layered nickelates.
2 Overview
2.1 Crystal Structure and Basic Chemistry
The copper oxide and layered d9 nickelate materials are transition metal oxide (TMO) compounds that share the common structural motif of the CuO2/NiO2 plane. We focus first on the conceptually simplest materials, “infinite layer” CaCuO2 and NdNiO2 [panel (A) of Figure 1] in which the transition metal ions occupy the vertices of a square planar array with the oxygen ions at the midpoints of the edges. In their bulk, three dimensional form these materials are stacks of transition metal/oxygen planes, with the Ca/Nd ions located half-way between planes, above the centers of the squares formed by four transition metal ions.
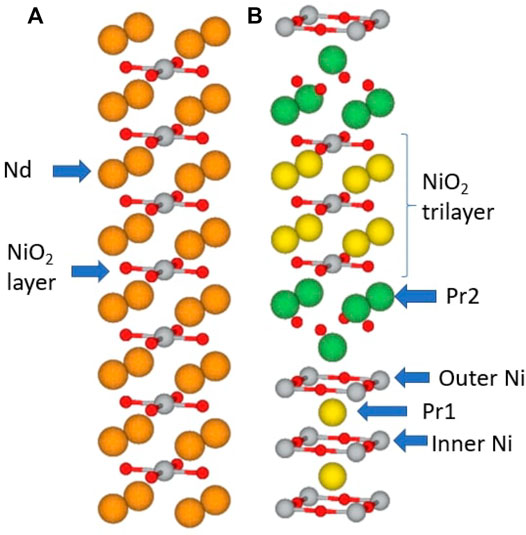
FIGURE 1. (A): crystal structure of infinite layer NdNiO2 in the P4/mmm structure. (B): crystal structure of trilayer Pr4Ni3O8 in the I4/mmm structure. Ni atoms are shown in silver, O in red, Nd in orange, Pr1 in yellow, and Pr2 in green. From Ref. [25] and the crystal structures are visualized using Vesta [115].
The CuO2/NiO2 plane motif may be combined in many ways, yielding a wide variety of compounds with somewhat similar properties but with many differences of detail. The very large number of cuprate materials will not be discussed here. In the nickelate materials, the infinite layer compounds can in principle be realized with all rare-earth elements R as A-site cation [26]. So far R = La, Pr, and Nd have been synthesized. In addition, multilayer variants R1+mNimO2m+2 are known, consisting of groups of m NdNiO2 planes separated by effectively insulating spacer layers of Nd and O. To date, m = 3, 4, 5 have been synthesized. The m = 3 material is shown in panel (B) of Figure 1; the plane labelled Pr2 is the spacer layer. Some charge transfer (CT) to the spacer layers occurs, leading to a doping of the NiO2 planes that is different than that of the infinite layer compounds, but apart from this the basic electronic physics of the layered compounds is believed to be very similar to that of the infinite layer compounds [27]. In this article we consider the compounds as interchangeable.
Qualitative chemical (“formal valence”) considerations suggest that the electronic configurations of the ions are Ca2+
Formal valence considerations are only a rough approximation to the actual electronic states because in a solid the charge on an ion is not conserved. Quantitatively or qualitatively important roles may be played by charge transfer processes including fluctuations that move an electron from an O to a transition metal (producing a
In the layered d9 nickelates such as NdNiO2 the situation is richer. Multiple bands cross the Fermi surface, while the potential relevance of d8 configurations raises the possibility that multiplet (“Hund’s”) interactions governing the relative energetics of different configurations of TMO d-electrons are relevant to the low energy physics. A fundamental question is whether this additional physics plays an important role in the low energy correlation physics such as superconductivity or whether the correlation physics of the nickelates is in essence similar to that of the cuprates.
2.2 Downfolding and the DFT + DMFT Methodology
In the context of quantum materials the process of defining an appropriately reduced model from a high energy, more chemically realistic, “all orbital” description is referred to as “downfolding”. Downfolding starts from the use of a mean field like approximation such as density functional theory to define a single-particle basis (e.g., the Kohn-Sham eigenstates within a broad energy region) that is for all practical purposes complete. Then a subspace of this set of states is selected and the Hamiltonian is appropriately projected onto the subspace. The required projection is more sophisticated than simply taking the matrix elements of the Hamiltonian between states in the subspace: screening of the retained interactions by processes involving states that are not retained must be incorporated and correlation induced shifts in relative energy splittings of different states (“double counting corrections”) must be managed correctly. Further, since the purpose is to obtain something that can be solved (perhaps approximately), the specifics of downfolding are intertwined with the method of solution of the resulting theory.
While one important goal of a downfolding process is to obtain a truly low energy theory (defined, say on the ±0.5 eV interval around the chemical potential that is relevant for analysis of transport and of low energy instabilities such as superconductivity), in the case of transition metal oxides an intermediate step of downfolding to a reduced model defined in a wider energy range but with truncated interactions has been found to be very useful. This intermediate step is motivated by the physical/chemical intuition that in transition metal oxides the most relevant interactions are the site local charging (“U”) and multiplet (“J”) interactions that control the energy levels of a partly filled d shell on a transition metal ion and by the success of the DMFT approximation in treating the resulting physics.
The DFT + DMFT methodology is a specific downfolding method based on the idea that both the important interactions and the correlation physics they induce are spatially local in a sense defined more precisely below. It is closely related to the DFT + U method inmpmllemented in standard DFT codes; indeed the DFT + U method is in effect the hartreee approximation to the DFT + DMFT method. The locality of the interactions greatly reduces the number of relevant interaction parameters (i.e., the complexity of the interacting Hamiltonian that must be solved) while the locality of the correlation physics drastically reduces the severity of entanglement effects, enabling a tractable solution of the resulting correlation problem. The method is now a work-horse of correlated electron materials science and the nickelate materials provide an interesting test of both the assumptions that go into the methodology and the results it produces.
The DFT + DMFT methodology [30, 31] uses a DFT calculation to define a set of correlated orbitals (for example the Ni d-orbitals), the hybridization of these orbitals to each other and to other orbitals, presumed to be uncorrelated, and also involves a prescription for associating site local interactions to the correlated orbitals. The resulting downfolded system may be thought of a kind of generalized multi-orbital Hubbard model involving correlated sites coupled to uncorrelated ones and is solved in the DMFT (locality of correlation physics) approximation which reduces the problem to the solution of a quantum impurity model (set of local orbitals coupled to a non-interacting bath) plus a self-consistency condition. Finally, in a charge self-consistent (CSC) step which is sometimes omitted, the full charge density of the system is obtained and fed back in to the density functional theory; the correlated orbitals are redefined, and the process is repeated until complete self consistency is obtained. This process is visualized in Figure 2. Importantly, the CSC DFT + DMFT equations can be derived as the stationarity conditions of a general functional describing the electronic structure ab-initio [31] within the DMFT approximations.
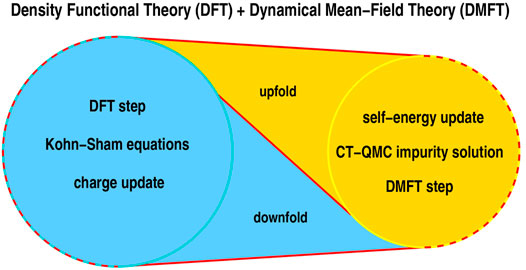
FIGURE 2. Principle DFT + DMFT method as hybrid scheme, working as a ratchet with up- and downfolding links. One starts from a converged KS-DFT calculation and downfolds to the correlated subspace where a DMFT step is performed. This asks mainly for the solution of a quantum-impurity problem [here conducted via continuous-time quantum-Monte Carlo (CT-QMC)], yielding a local self-energy. That self-energy is upfolded to the Bloch space of the DFT problem and used to revise the electronic charge density. Using the latter, a new KS potential is generated and a novel DFT step performed, which is followed by the next downfolding, etc. This cycle is performed until self-consistency in the charge density and the self-energy is achieved. Adapted from Ref. [116].
In first instance, DMFT delivers results for the electron (one-particle) Green’s function
From solving the impurity problem within DMFT the atomic multiplet nature of the correlated manifold can also be analyzed from the many-body density matrix. The computed spectral functions can be compared directly to photoemission measurements [32, 33] and considerable intuition about the physics can be gained from the many body density matrices. Furthermore, symmetry broken phases such as superconductivity and magnetic phase can be addressed, and transport coefficients can be estimated. Momentum dependent susceptibilities and vertex functions can also be constructed, albeit with much more computational effort.
The important technical steps in implementing the dynamical mean field approximation are the construction of the correlated orbitals and their coupling to the uncorrelated orbitals and the computation of the interactions among electrons in these orbitals.
3 Methods
The DFT + DMFT methodology is formulated in terms of the single particle Green function, defined on the Matsubara (imaginary frequency) axis for a periodic solid as
Here k is a wavevector in the first Brillouin zone of the solid, μ is the chemical potential, Href is a reference single-particle (non-interacting) Hamiltonian and the self energy operator
Here, we take Href to be the Kohn-Sham Hamiltonian produced by a specified density functional theory combined with a “double counting (DC) correction” and
In embedding theories such as DFT + DMFT one defines on physical grounds a subset of correlated orbitals (in the cuprate/nickelate context the transition metal d-orbitals) and retains only the site-local matrix elements of the self energy among these orbitals and the site-local interactions that contribute to these matrix elements. These site-local self energy matrix elements are then calculated via a mapping to a quantum impurity model with the local Hamiltonian (crystal field levels plus interactions) of the full model and a self-consistently determined coupling to a bath in the form of a so called “hybridization function”; the result is self-consistently embedded into the full electronic structure.
The key conceptual issues in the method are the construction of the local orbitals and the specification of the interactions between them. These issues are discussed in the next two subsections.
3.1 Quantum Embedding: Construction of the Localized Orbitals
The first step in this procedure is to precisely define a set of orbitals
which specify the relation of the local orbitals
The projector functions allow one to “upfold” a self energy calculated in the orbital (Ri, m) basis to the Kohn-Sham basis as
Here we have written Eq. 4 for the DMFT ansatz of a self energy that is site local in the orbital basis. In general the self-energy can also contain multiple sites embedded as a cluster impurity. However, in this case one has to carefully consider breaking of symmetries due to the geometry of the lattice.
The full Green function Eq. 2, a matrix in the set of Kohn-Sham bands, is then written as
The ‘downfolded’ site-local lattice Green function
which allows to construct the dynamic Weiss field of the quantum impurity problem in DMFT via a Dyson equation:
Solving the impurity problem gives a new self-energy (Eq. 4), defining a new lattice Green function, and hence a new local Green function. This procedure is performed until convergence is reached and
The DMFT converged lattice Green function Eq. 5 can be used to construct a new charge density:
which serves as input for DFT in a CSC DFT + DMFT calculation. From this a new charge density from DMFT a new set of Kohn-Sham (KS) orbitals, projectors and interactions are defined and the DMFT equations are solved again. This procedure is repeated until the charge density converges. The effect of this additional charge self consistency loop is sometimes small, but in many cases can have important quantitative influence in the results [36], and is technically needed to evaluate energies within the DFT + DMFT formalism [31].
3.1.1 Projected Atomic Orbitals
One choice for constructing the projector functions is the projected atomic orbitals (PLO) methodology [37–39], in which one introduces a set of atomic-like correlated orbitals
The downfolded orbitals describing the correlated subspace are computed as
here,
The window
3.1.2 Maximally Localized Wannier Functions for Downfolding
One may also obtain the projector functions via the Wannier construction introduced by Marzari and Vanderbilt [40], in which one defines N spatially localized Wannier functions (WFs) as Fourier transforms of linear combinations of N KS states via
Here the
A subset of the Wannier functions, labelled by m, are identified with the correlated orbitals and the Wannier construction in Eq. 11 implies that projectors can be identified as
This construction is implemented in the software package Wannier90 (W90) [42].
The initial seed for
The number of Kohn-Sham bands NB used in the Wannierization process can be chosen similar to the projector method above via a window
This ensures “global smoothness of connection” and an optimal k-point connectivity by minimizing the gauge invariant term of the spread of the resulting WFs [43]. Afterwards, the spread-minimization is performed as usual using the Ψ(opt). This allows to define the projector functions as:
It is important to note that while the projection of the KS Hamiltonian onto a given set of Wannier functions may reproduce the Kohn-Sham bands perfectly, different Wannierization choices may lead to different constructions of the orbitals and to different tight binding parameters. This issue is discussed in more detail in Ref. [44].
3.1.3 Comparison
The projector and Wannier constructions of the localized orbitals are conceptually very similar, differing in the specifics of how the correlated orbitals are constructed. The projector method is connected in a intuitively appealing manner to the local orbitals of interest (especially if the projection window
The MLWF method is less dependent on an a-priori choice of atomic orbitals, is based on minimization of a clear metric under which the orbitals are constructed, and also minimizes deviations from the KS dispersion. Because it constructs a basis set which is complete within some energy range, it provides at no additional complication a full tight-binding parametrization of the electronic band structure in a given energy range,
providing physical intuition about the relevant orbitals. (With some effort, similar information can be obtained from the projector method, but this is not often done). Figure 3 shows two examples of fitting the NdNiO2 low-energy KS states using MLWF. The first [Figure 3A], constructs 17 Wannier orbitals for all 17 KS states that are present in a large energy window from -9 to 8 eV, whereas the second [Figure 3B] constructs a minimal model only for the three frontier orbitals (Ni-
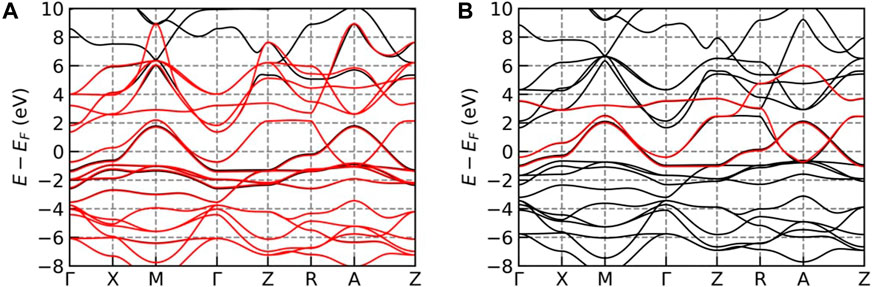
FIGURE 3. DFT bands for NdNiO2 (black lines) and Winner fits (red lines) for Wannierization using (A) 17 Wannier functions (5 Nd-d orbitals +5 Ni-d orbitals + 6 oxygen-p orbitals + interstitial-s orbital and (B) using 3 Wannier functions (Nd-
In the following we will call all calculations performed directly in the tight-binding (TB) basis Wannier-mode calculations, whereas calculations in which the DMFT equations are written in the Kohn-Sham basis will be referred to as Bloch-mode calculations. Note, that direct formulation of the DMFT equations as a solution of a Hamiltonian restricted to the space of the correlated orbitals is in general not possible, because the projectors are in general non-square matrices and hence do not serve as a unitary transformation [46].
While Wannier and projector methods had until recently been viewed as roughly equivalent methods of constructing the basis in which the dynamical mean field equations are formulated, and indeed in some cases the equivalence of projector and Wannier-based methods was demonstrated [47], Karp et al. [44] found that results of the DFT + DMFT methodology can be sensitive to the choice of method used to construct the local orbitals of the downfolded model. We will review and discuss these results in Section 4.2.
3.2 Interactions in the Correlated Subspace
3.2.1 Basic Definitions
Once an orbital downfolding has been defined one has to formulate an appropriate interaction among the downfolded correlated orbitals
Here,
In the absence of symmetries there are
The most widely used form is the so called “Slater” parametrization [49], which makes use of the spherical symmetry of an isolated single atom. If the projectors in Eq. 3 are chosen to produce sufficiently local WFs this is a good approximation, and is for example used in all DFT + U implementations.
The most important Coulomb integrals are elements of
Here, Fk, refers to the kth Slater integral, proportional in effect to the kth-pole of the electric charge distribution of the atomic configuration of the rotationally symmetric free-ion case. From the Fk the full Coulomb tensor can be constructed. For a d-shell of an isolated ion one can further show that
and that F4/F2 is fixed, so that the entire Coulomb interaction tensor is determined by only two parameters: F0 = Uavg and Javg. The ratio F4/F2 is obtained empirically, varies only little for transition metals, and it is often fixed to F4/F2 ≈ 0.625 [50].
Another often applied parametrization is the so called “Hubbard-Kanamori” form [51, 52], widely used in particular to describe the on-site interactions among electrons in the d-shell of a transition metal ion. This parametrization is formulated in terms of the averaged parameters:
which are the so-called Hubbard-Kanamori parameters for n orbitals. In the particular case of an octahedral crystal field, the d shell is split into a t2g and eg manifold. Within either the t2g and eg sub-manifold of the octahedral point group (but not for the whole d shell), one can verify that
Importantly, this form of the interaction is rotationally invariant, which means that within the subset of orbitals, arbitrary unitary transformations on the orbitals can be applied, without adapting the parameters.
Note, that in this form
3.2.2 Determining the Screened Interactions
In solids the interaction parameters are renormalized from their bare values by screening processes V → U involving the other electrons in the solid. The most important renormalization is of the monopole interaction F0 ≡ Uavg which gives the charging energy
Commonly used ab-initio methods for treating the strong renormalizations from solid state screening are the constrained LDA method [53] and the constrained random phase approximation (cRPA) method [54]. Here, we present the latter, for a review see e.g., Ref. [55].
cRPA is a linear response theory based on the polarization function P giving the response of electrons to a test charge in the system. Within the RPA approximation, which neglects all non-Hartree terms, the dielectric function is calculated from the full bare Coulomb interaction and the polarization function as:
The polarization function P in RPA is calculated from DFT as:
where Ψνk and ϵνk mark KS eigenstates and eigenvalues.
The effective screened Coulomb interaction U in a target “(t)” space (typically the manifold of correlated states)can now be calculated as follows. Consider the example depicted in Figure 4. The effective screened Coulomb interaction is calculated by first splitting the polarization of the system in two parts, Pt pertaining only to transitions among the target orbitals and Pr containing all other transitions (including from target to non-target orbitals and the reverse):
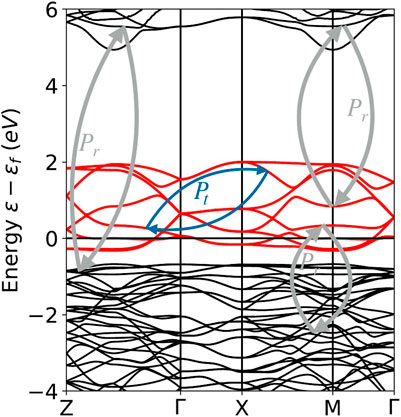
FIGURE 4. Band structure of LuNiO3, with constructed Wannier functions for the Ni eg states as target states t. The decomposition in the polarization channels Pt within the correlated subspace, and the polarization channels Pr outside, from, and to the target subspace t are schematically depicted as blue and grey arrows.
Now one can deduce the partially screened interaction Wr from Pr as:
Here Wr, which is implicitly restricted to include only matrix elements among the target orbitals, is Coulomb interaction tensor
Note, that due to the energy dependency of the polarization, Wr is naturally frequency dependent. The frequency dependence is often neglected and U (ω = 0) is used.
Now, one can analyze the symmetries of the calculated Coulomb tensor and fit to one of the two forms given above. Either by directly averaging the elements of the tensor, or using a minimization procedure to minimize differences between UcRPA and a constructed U. Importantly, U should be evaluated in the same orbitals used for the embedding techniques. We advocate the use of a more advanced version of cRPA for disentangled bands implemented in VASP evaluating the polarization directly using the WFs [56].
It is evident that the screening depends crucially on the chosen subspace. For example, a larger energy window for the target space in the downfolding produces more atomic like orbitals but also has fewer screening channels, so leads to a larger interaction, whereas smaller energy windows give more extended orbitals with smaller, heavily screened, interactions. As we will show later in Section 4.2 the screening in infinite layer nickelates is very sensitive as both, oxygen p states below, and Nd d states above, make large contributions to the screening. We discuss results from cRPA in Section 4.3.
3.3 Including Local Coulomb Interactions on Oxygen: DFT + sicDMFT Approach
In standard DFT + DMFT the many-body physics imposed by U on Ni is treated within the DMFT correlated subspace, however the description of the quantum processes on O remains on the Kohn-Sham DFT level. Note that correlations on the O (2p) orbitals are not necessarily weak because these orbitals, just as Ni(3d) ones, carry the first new angular-momentum number with growing main quantum number (there is neither a 1p nor a 2d orbital), meaning the orbitals may sit close to the atomic nucleus and therefore are more compact and with a larger charging energy. This implies that also 2p frontier orbitals have a demanding pseudopotential that needs to moderate between localization and itinerancy (though not as severely as for 3d orbitals). And this nature becomes increasingly relevant when the connection to the environment via a Δ comparable to U is substantial, a feature taking place for later TMOs. This suggests that a DFT-level treatment may be insufficient to tackle the sophisticated Mott-Hubbard vs. charge-transfer balance. As a further aspect in infinite-layer nickelates, while Ni-
However, treating explicit Coulomb interaction within DMFT also for O(2p) raises several methodological and numerical challenges, and the explicit quantum-fluctuating aspect in these orbitals should be still generally weaker than in transition-metal 3d orbitals. In order to include localization tendencies on the oxygen sites beyond KS-DFT, one may therefore introduce the self-interaction correction (SIC) [34] as a simplified treatment compared to DMFT. The SIC scheme can efficiently be applied already on the pseudopotential level [57–59], leading to a refined O pseudopotential to be used in a standard CSC DFT + DMFT [60]. Use of this pseudopotential defines the DFT + sicDMFT method, which is thus capable of handling correlation physics on and between Ni and O closer to equal footing.
Figures 5A,B show the principal impact of the SIC inclusion on the DFT level. We see that the radial part of the O (2p) pseudopotential is somewhat reduced within the lower-limit of the bonding region (∼ 0.5–2 a.u.), resulting in a stronger localization of charge near the O site. For the NdNiO2 crystal calculation within DFT + sic, i.e., employing the revised oxygen pseudopotential in the conventional KS cycle, two key effects may be observed. First, the O(2p) block of six bands (originally between
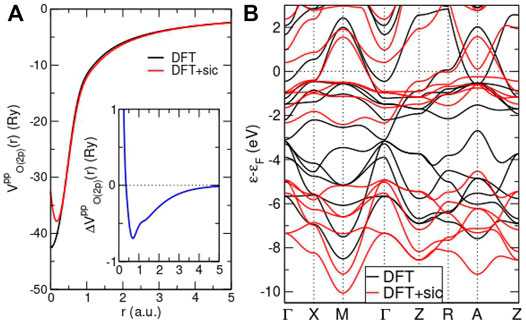
FIGURE 5. Effects of SIC inclusion on the DFT level. (A) Radial part of oxygen 2p pseudopotential with (red) and without SIC (black). Inset: difference between DFT and DFT + sic pseudopotential. (B) Band structure for NdNiO2 within DFT (black) and DFT + sic (red). Note that in the latter calculation, SIC was only applied to O and the Ni orbitals are treated on the DFT level. F.L. to be published.
4 Results
In this section, we give an overview of results on infinite-layer nickelates in literature and a comparison to the better understood case of the layered copper oxides. We mainly focus on theoretical results [12, 13, 15–19, 21, 23, 27, 33, 44, 61–85] but present some comparison to experiments [4, 10, 86–98], when the relevant experimental results are available. We also mention that while we are aware of the important works on the study of interface effects [63, 65, 69], due to space limitation, we concentrate on the study of bulk nickelates here.
We present four levels of results: for orientation we show the DFT-level electronic structure; then we describe the basic many-body electronic structure following from the different DFT + DMFT calculations and the approximate physical picture that results. Next we consider the predictions of these calculations for the Fermiology–which bands are present at the Fermi surface and what are the mass enhancements. Finally, inspired by recent experimental results, we make some brief remarks about magnetism in Section 4.4.
4.1 DFT Results
Figure 6 compares basic aspects of the DFT-level electronic structure of the infinite layer nickelates NdNiO2 and the analogous cuprate compound CaCuO2. Panels a and d present the DFT bands of the two compounds. Panel d shows the familiar cuprate band structure, with one essentially two dimensional band of mixed Cu-
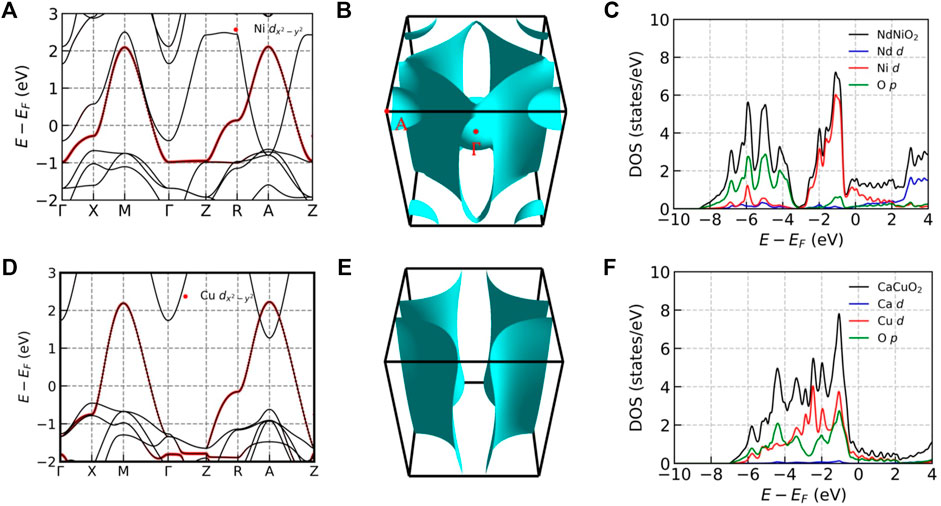
FIGURE 6. (A–C): Electronic properties of NdNiO2. (A): Electronic band structure of NdNiO2 close to the Fermi level. The red dots highlight the Ni
Charge transfer from the
It is important to note that the published DFT analyses of orbital admixture are obtained by projecting the states onto atomic orbitals as described in the projector section above. Gu et al. [21] find via a Wannier analysis that the additional band also has considerable contribution from charge density not centered on any atom. Because this component is not centered on an atom it is not easily revealed in the standard projector analysis. Panel a of Figure 7 shows this component, known as an interstitial s orbital, which is located at the mid-point between two neighboring Ni atoms along the z axis. Panels b and c show the fitting of DFT band structure using maximally localized Wannier functions (MLWF) as explained in the previous Methods section. Panel b shows a band fit based on 16 MLWFs, of which 5 are initialized as being centered on Ni-d orbitals, five more centered on Nd-d orbitals and six O-p orbitals. The fitting is very good for the occupied bands but for one of the Nd-derived empty bands, the Z → R portion is not well reproduced. Panel c shows the result of adding one more MLWF that corresponds to the interstitial s orbital. The fitting is improved, in particular in that Nd-derived bands are now exactly reproduced throughout the first Brillouin zone. Panel d shows the weight of the interstitial s orbital on the different bands. We can see that the s-orbital has a high weight on the extra band on the Z → R portion at energy E ≈ 2 eV above the Fermi level.
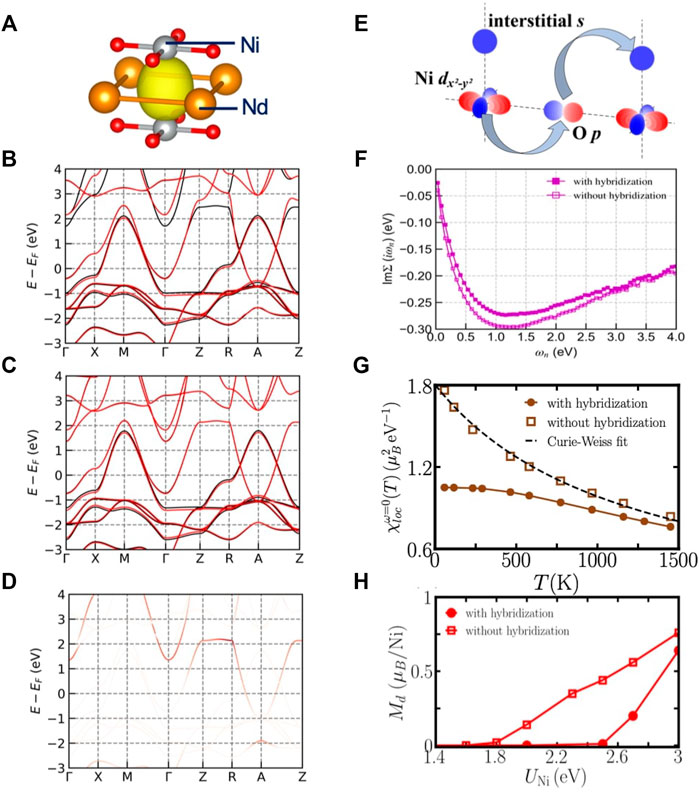
FIGURE 7. (A): An iso-value surface of the interstitial s orbital in NdNiO2. (B): Fitting of DFT band structure of NdNiO2, using 16 Wannier functions (Nd-d, Ni-d, and O-p orbitals). The black lines are DFT bands and the red lines are fitted bands from Wannier functions. (C): Fitting of DFT band structure of NdNiO2, using 17 Wannier functions (Nd-d, Ni-d, O-p, and the interstitial s orbitals). The black and red lines have the same meaning as in panel (B). (D): The fatband plot of the interstitial s orbital. (E): An illustration of the hybridization between Ni
One of the key questions in the materials physics of the layered d9 nickelates is whether the additional band is a “spectator”, acting simply as a reservoir enabling charge transfer from the NiO2 plane to the Nd spacer layer, or whether the additional band also plays an essential role in the physics, either because it is strongly hybridized with or strongly interacting with the Ni degrees of freedom. The orbital composition of the additional band is relevant to this question: the “spectator band” contains Nd
To complete the discussion of DFT-level theory we mention DFT + U calculations on infinite-layer nickelates. In these calculations all the atomic orbitals that are in the pseudo-potentials are taken into account [11, 24, 26] and rotationally invariant Hubbard U interactions are added on all the five Ni d orbitals. Botana et al. [24] extracted various hopping matrices and energy splitting, which shows similarity between infinite-layer nickelates and cuprates. Kapeghian et al. [26], Been et al. [11] and Xia et al. [104] studied the electronic structure trends of the entire lanthanide series of infinite-layer nickelates.
4.2 DFT + DMFT: Local Electronic Structure
In transition metal oxides, it is believed that the interesting correlation physics arises from a competition between local interactions within the transition metal d-shell, which control the relative energetics of different d-multiplets, and the hybridization with other orbitals, which acts to mix the d-multiplets. In assessing the relevance of different interactions, an analysis of the ground state wave function is of interest. As noted previously, a formal valence analysis places either the Ni or the Cu in a d9 state, thus with one hole in the d-shell and full oxygen-2p and empty Nd-5d/6s shells. Deviations from this simple picture provide insight into the relevant interaction processes. One question is the admixture of ligand (O-2p holes in the cuprate and nickelate cases and also Nd-5d/6s electrons in the nickelate case) states. One distinguishes [105] “charge transfer” materials where the energy difference between the ligand and transition metal d states controls the physics from Mott Hubbard materials where the charging energy of the transition metal d-shells controls the physics. A second issue is the relative weight of different transition metal multiplets. In “Hund’s metals”, multiplet configurations involving high spin (spin S ≥ 1) d-states are relevant; in Mott Hubbard materials only the S = 1/2 and S = 0 states are relevant.
DFT + DMFT calculations provide theoretical estimates of orbital occupancies and of the local density matrices describing the multiplet probabilities of the correlated sites and the occupancies of the ligand sites. In the cuprate case there is general agreement both within DFT and in DFT + DMFT that the only relevant states are d9 (with the hole in the Cu-
In the nickelate compounds the theoretical situation is less clear. There is a general consensus that the Ni-
Perhaps more importantly, unlike the cuprate case the transition metal Ni-
These questions are not theoretically settled because, as shown by Karp et al. [44] the choice of method used to construct the local orbitals of the downfolded model affects the DMFT results for the nickelate. Table 1 compares the orbital occupancies and multiplet occurence probabilities (defined as weight of the different configurations in the many-body density matrix projected onto the Ni states) obtained with different methodologies.
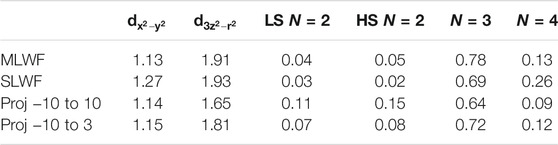
TABLE 1. Orbital occupancies of the most relevant Ni-d orbitals of NdNiO2 from the Matsubara Green function (left) and occurrence probabilities of low spin (LS S = 0) and high spin (HS S = 1) multiplet configurations obtained from the impurity density matrix computed for stoichiometric NdNiO2 using a Kanamori Hamiltonian with two correlated orbitals and
4.3 DMFT Theory of the Low Energy Physics
We next consider the consequences of the wide window electronic structure for the low energy physics. Panels (a) and (b) of Figure 8 show the many body electronic structure (momentum and frequency dependent electron spectral function) computed for NdNiO2 at two representative dopings using the basic DFT + DMFT and DFT + sicDMFT methods for a particular method and choice of parameters. From a comparison with other nickelates [60] and in view of experimental constraints (see discussion in [106]) a value Uavg = 10 eV and Javg = 1 eV is here used to parametrize the local Coulomb interaction for the Ni(3d) orbitals in charge self-consistent DFT+(sic)DMFT calculations. The projector method for the five Ni(3d) orbitals, building on the 12 KS states above the O(2s) bands [i.e., an energy window (−10, 3) eV], is employed and a rotational-invariant Slater Hamiltonian is active in the resulting correlated subspace. The hole doping δ is achieved by the virtual-crystal approximation using an effective Nd atom [108], where the Nd (4f) states are frozen in the pseudopotential core. Note again that calculational settings resulting in the data shown in Figure 8 differ only in using the LDA (SIC) oxygen pseudpotential in DFT+(sic)DMFT.
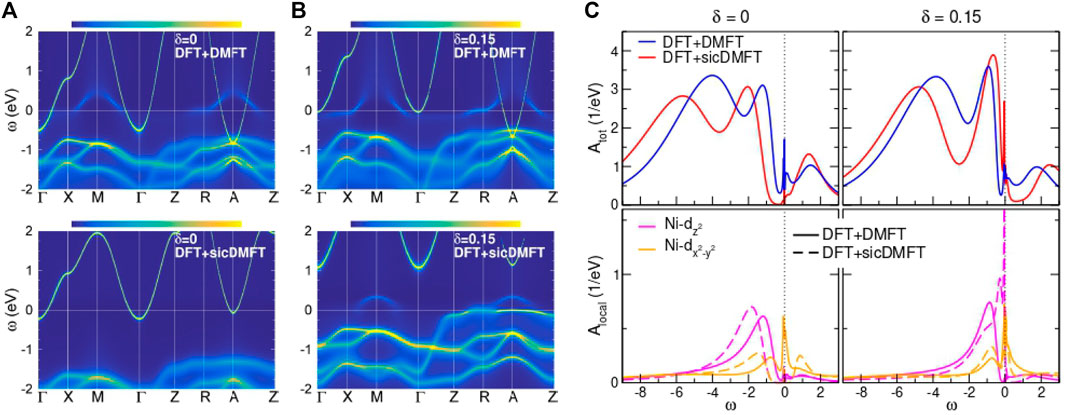
FIGURE 8. Comparison between DFT + DMFT and DFT + sicDMFT at T = 30 K, using Uavg = 10 eV, Javg = 1 eV and projected-local orbitals on the 12 KS states above the O(2s) bands for both schemes, respectively. (A,B) k-resolved spectral function A (k, ω) for (A) pristine and (B) δ = 0.15 hole-doped NdNiO2. (C) Total and Ni-eg local spectral function for both doping cases. Adapted from [108].
The upper panels of Figures 8A,B show the DFT + DMFT spectral function. In comparison to the DFT bands shown in Fig. 6, we see that the Ni-
As depicted in Figure 9, the increase of correlation strength with SIC inclusion originates from the stronger localization of O(2p) electrons. The oxygen states are shifted down in energy, leading to an increase of the p-d splitting and thus to a value Δ = 5 eV for the charge-transfer energy [106]. Figure 9B shows directly in real space, that the SIC-modified pseudopotential of oxygen enhances the 2p charge density around the oxygen sites, with additionally further depleting Ni-
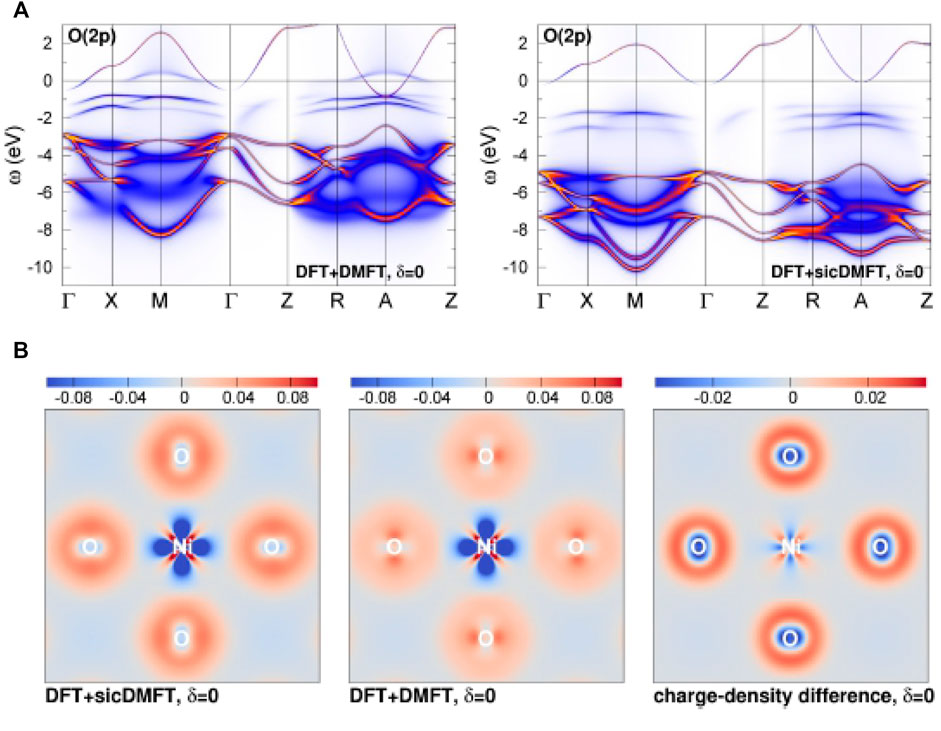
FIGURE 9. Oxygen 2p states in DFT + DMFT and DFT + sicDMFT. (A) Orbital weight (i.e., fatbands) in the interacting regime along high symmetry lines. (B) Interacting bond charge density
These results highlight the basic electronic structure questions: 1) what is the coherence (scattering rate and mass enhancement) of the Ni-
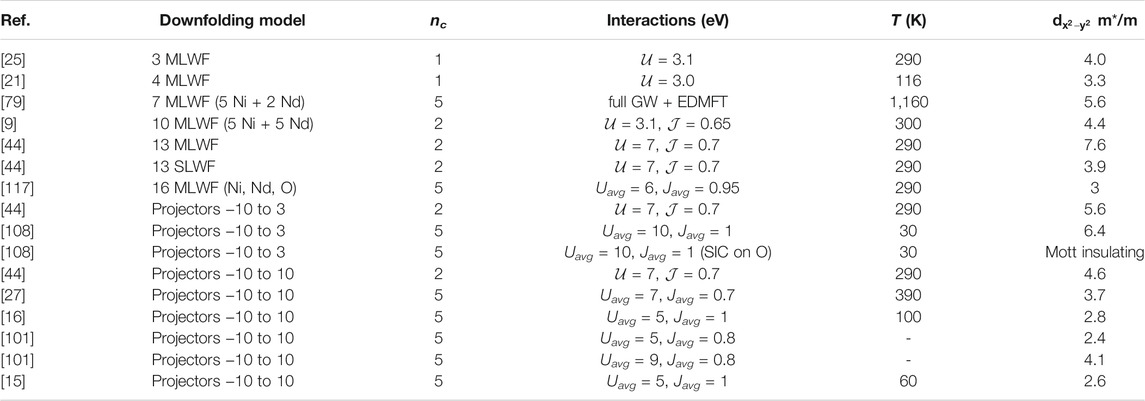
TABLE 2.
The question of band renormalization in different downfolding choices is tightly coupled with the question of screening (see Section 3.2.2). cRPA studies showed that if a seven orbital downfolding model is constructed from five Ni(3d) orbitals and two Nd(5d) orbitals in a small energy window (not containing any oxygen states), the static part of the onsite Coulomb interaction is
Finally, we may consider the functional form of the self energy, which is relevant to the issue of Hund’s physics. Hund’s metal physics is believed to imply a particle-hole asymmetric structure in the self energy leading to an extra peak in the electron spectral function [33, 109]; for materials such as NdNiO2 where the d-shell is more than half filled the peak is on the unoccupied part of the spectrum [33]. Conversely, Mott physics would result in a two peak spectral function and a more symmetric self energy with peaks on both sides. As shown in Figure 10, the
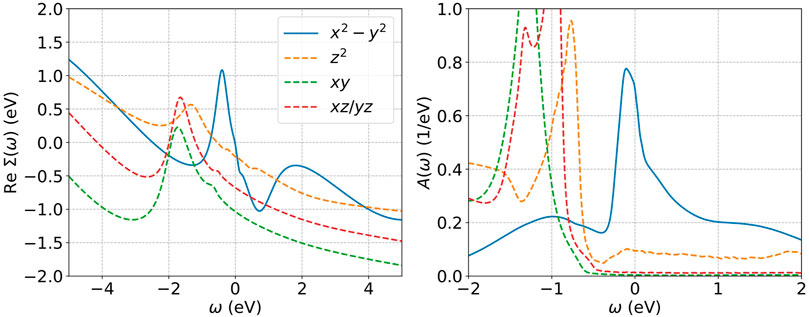
FIGURE 10. Left: Self energy of the different orbitals in a five orbital fully charge self consistent DFT + DMFT calculation at stoichiometry with projectors in an energy range from −10 to 10 eV around the Fermi level, using a rotationally invariant slater Hamiltonian with Uavg = 7 eV, Javg = 0.7 eV, and T = 290K. Right: corresponding momentum integrated spectral function. Adapted from Ref. [27].
4.4 Magnetism
We now discuss briefly the magnetic properties. The stoichiometric cuprates are antiferromagnetic insulators, with a charge gap of approximately 1.5 eV, a Neel temperature
A discussion of magnetism in the nickelates is complicated by the self-doping effect. If the physics of the nickelates is directly comparable to that of the cuprates then one would expect the stoichiometric nickelates to be on the boundary of magnetism. Recent resonant inelastic X-ray (RIXS) [92] and nuclear magnetic resonance experiments [91] are consistent with this picture. In particular, Lu et al. report no long ranged order but observe a strong zone boundary paramagnon-like excitation implying a JNN ≈ 60 meV, about half of the cuprate value.
Theoretically a number of DFT + U, hybrid DFT and DFT + DMFT studies have studied magnetism of infinite layer nickelates [17, 21, 24, 25, 84, 99, 101, 108, 111] and reported a wide range of magnetic superexchange [17, 18, 23, 66, 76, 77, 99, 112] from a small value of about 10 meV [23, 99] to an intermediate value of about 30 meV [66] to a large value of about 80–100 meV [17, 18, 76, 77, 112]. DFT + DMFT studies have not examined the Ni exchange coupling systematically. Further investigation of the magnetic properties within this method is likely to provide valuable insights.
5 Summary
DFT and beyond DFT analyses have produced a broadly coherent picture of the electronic structure of the infinite layer d9 nickelates. Similar to the cuprates there is a rather two dimensional Ni-
Given this broad consensus, the question becomes which of the differences and similarities to the cuprates are important for the low energy physics. It is clear that the additional band “self-dopes” the Ni
In this article we have explained the different choices and the different results that emerge. Some of the differences in results are experimentally testable. For example, the DFT + (sic)DMFT approach yield significantly more strongly correlated/less coherent
We hope that our work will motivate comparisons to experiment and to more fundamental theoretical approaches that will help resolve some of the methodological questions relating to the DFT + DMFT approach to correlated materials.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
HC is supported by the National Natural Science Foundation of China under project number 11 774 236, the Ministry of Science and Technology of China under project number SQ2020YFE010418, and NYU University Research Challenge Fund. FL and JK were supported by grants from the Simons Foundation. The Flatiron Institute is a division of the Simons Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
HC acknowledges useful discussions with Yuhao Gu, Mi Jiang, Danfeng Li, Jiawei Mei, Xiangang Wan, Tao Wu, Xianxin Wu, Yifeng Yang, Guangming Zhang, Zhicheng Zhong, Wei Zhu, and in particular, Chengliang Xia for his help in making part of the figures.
References
1. Bednorz JG, Miller KA. Possible highT C Superconductivity in the Ba-La-Cu-O System. Z Physik B - Condensed Matter (1986) 64:189–93. doi:10.1007/BF01303701
2. Wu MK, Ashburn JR, Torng CJ, Hor PH, Meng RL, Gao L, et al. Superconductivity at 93 K in a New Mixed-phase Y-Ba-Cu-O Compound System at Ambient Pressure. Phys Rev Lett (1987) 58:908–10. doi:10.1103/PhysRevLett.58.908
3. Orenstein J, Millis AJ. Advances in the Physics of High-Temperature Superconductivity. Science (2000) 288:468–74. doi:10.1126/science.288.5465.468
4. Li D, Lee K, Wang BY, Osada M, Crossley S, Lee HR, et al. Superconductivity in an Infinite-Layer Nickelate. Nature (2019) 572:624–7. doi:10.1038/s41586-019-1496-5
5. Anisimov VI, Bukhvalov D, Rice TM. Electronic Structure of Possible Nickelate Analogs to the Cuprates. Phys Rev B (1999) 59:7901–6. doi:10.1103/PhysRevB.59.7901
6. LeBlanc JPF, Antipov AE, Becca F, Bulik IW, Chan GK-L, Chung C-M, et al. Solutions of the Two-Dimensional hubbard Model: Benchmarks and Results from a Wide Range of Numerical Algorithms. Phys Rev X (2015) 5:041041. doi:10.1103/PhysRevX.5.041041
7. Zheng B-X, Chung C-M, Corboz P, Ehlers G, Qin M-P, Noack RM, et al. Stripe Order in the Underdoped Region of the Two-Dimensional hubbard Model. Science (2017) 358:1155–60. doi:10.1126/science.aam7127
8. Jiang H-C, Devereaux TP. Superconductivity in the Doped Hubbard Model and its Interplay with Next-Nearest Hopping T ′. Science (2019) 365:1424–8. doi:10.1126/science.aal5304
9. Kitatani M, Si L, Janson O, Arita R, Zhong Z, Held K. Nickelate Superconductors-A Renaissance of the One-Band Hubbard Model. Npj Quan Mater. (2020) 5:59. doi:10.1038/s41535-020-00260-y
10. Hepting M, Li D, Jia CJ, Lu H, Paris E, Tseng Y, et al. Electronic Structure of the Parent Compound of Superconducting Infinite-Layer Nickelates. Nat Mater (2020) 19:381–5. doi:10.1038/s41563-019-0585-z
11. Been E, Lee WS, Hwang HY, Cui Y, Zaanen J, Devereaux T, et al. Electronic Structure Trends across the Rare-Earth Series in Superconducting Infinite-Layer Nickelates. Phys Rev X (2021) 11:011050. doi:10.1103/physrevx.11.011050
12. Hu L-H, Wu C. Two-band Model for Magnetism and Superconductivity in Nickelates. Phys Rev Res (2019) 1:032046. doi:10.1103/PhysRevResearch.1.032046
13. Werner P, Hoshino S. Nickelate Superconductors: Multiorbital Nature and Spin Freezing. Phys Rev B (2020) 101:041104. doi:10.1103/PhysRevB.101.041104
14. Zhang Y-H, Vishwanath A. Type-II t−J Model in Superconducting Nickelate Nd1−xSrxNiO2. Phys Rev Res (2020) 2:023112. doi:10.1103/PhysRevResearch.2.023112
15. Kang C-J, Kotliar G. Optical Properties of the Infinite-Layer La1−xSrxNiO2 and Hidden Hund's Physics. Phys Rev Lett (2021) 126:127401. doi:10.1103/PhysRevLett.126.127401
16. Wang Y, Kang C-J, Miao H, Kotliar G. Hund's Metal Physics: From SrNiO2 to LaNiO2. Phys Rev B (2020) 102:161118. doi:10.1103/PhysRevB.102.161118
17. Wan X, Ivanov V, Resta G, Leonov I, Savrasov SY. Exchange Interactions and Sensitivity of the Ni Two-Hole Spin State to Hund's Coupling in Doped NdNiO2. Phys Rev B (2021) 103:075123. doi:10.1103/PhysRevB.103.075123
18. Wu X, Di Sante D, Schwemmer T, Hanke W, Hwang HY, Raghu S, et al. Robust Dx2−y2 -wave Superconductivity of Infinite-Layer Nickelates-Wave Superconductivity of Infinite-Layer Nickelates$d_{x^{2}-Y^{2}}$fx169. Phys Rev B (2020) 101:060504. doi:10.1103/PhysRevB.101.060504
19. Nomura Y, Hirayama M, Tadano T, Yoshimoto Y, Nakamura K, Arita R. Formation of a Two-Dimensional Single-Component Correlated Electron System and Band Engineering in the Nickelate Superconductor NdNiO2. Phys Rev B (2019) 100:205138. doi:10.1103/PhysRevB.100.205138
20. Lechermann F. Multiorbital Processes Rule the Nd1−xSrxNiO2 Normal State. Phys Rev X (2020) 10:041002. doi:10.1103/PhysRevX.10.041002
21. Gu Y, Zhu S, Wang X, Hu J, Chen H. A Substantial Hybridization between Correlated Ni-D Orbital and Itinerant Electrons in Infinite-Layer Nickelates. Commun Phys (2020) 3:84. doi:10.1038/s42005-020-0347-x
22. Gao J, Peng S, Wang Z, Fang C, Weng H. Electronic Structures and Topological Properties in Nickelates Ln n+1NinO2n+2. Natl Sci Rev (2021) 8. doi:10.1093/nsr/nwaa218
23. Jiang M, Berciu M, Sawatzky GA. Critical Nature of the Ni Spin State in Doped NdNiO2. Phys Rev Lett (2020) 124:207004. doi:10.1103/PhysRevLett.124.207004
24. Botana AS, Norman MR. Similarities and Differences between LaNiO2 and CaCuO2 and Implications for Superconductivity. Phys Rev X (2020) 10:011024. doi:10.1103/PhysRevX.10.011024
25. Karp J, Botana AS, Norman MR, Park H, Zingl M, Millis A. Many-body Electronic Structure of NdNiO2 and CaCuO2. Phys Rev X (2020) 10:021061. doi:10.1103/PhysRevX.10.021061
26. Kapeghian J, Botana AS. Electronic Structure and Magnetism in Infinite-Layer Nickelates RNiO2 ( R=La−Lu ). Phys Rev B (2020) 102:205130. doi:10.1103/PhysRevB.102.205130
27. Karp J, Hampel A, Zingl M, Botana AS, Park H, Norman MR, et al. Comparative many-body Study of Pr4Ni3O8 and NdNiO2. Phys Rev B (2020) 102:245130. doi:10.1103/PhysRevB.102.245130
28. Park H, Millis AJ, Marianetti CA. Site-selective mott Transition in Rare-Earth-Element Nickelates. Phys Rev Lett (2012) 109:156402. doi:10.1103/PhysRevLett.109.156402
29. Mizokawa T, Khomskii DI, Sawatzky GA. Spin and Charge Ordering in Self-Doped mott Insulators. Phys Rev B (2000) 61:11263–6. doi:10.1103/PhysRevB.61.11263
30. Georges A. Strongly Correlated Electron Materials: Dynamical Mean-Field Theory and Electronic Structure. AIP Conf Proc (2004) 715:3–74. doi:10.1063/1.1800733
31. Kotliar G, Savrasov SY, Haule K, Oudovenko VS, Parcollet O, Marianetti CA. Electronic Structure Calculations with Dynamical Mean-Field Theory. Rev Mod Phys (2006) 78:865–951. doi:10.1103/RevModPhys.78.865
32. Tamai A, Zingl M, Rozbicki E, Cappelli E, Riccò S, de la Torre A, et al. High-Resolution Photoemission on Sr2RuO4 Reveals Correlation-Enhanced Effective Spin-Orbit Coupling and Dominantly Local Self-Energies. Phys Rev X (2019) 9:021048. doi:10.1103/PhysRevX.9.021048
33. Karp J, Bramberger M, Grundner M, Schollwöck U, Millis AJ, Zingl M. Sr2MoO4 and Sr2RuO4 : Disentangling the Roles of Hund's and Van Hove Physics. Phys Rev Lett (2020) 125:166401. doi:10.1103/physrevlett.125.166401
34. Perdew JP, Zunger A. Self-interaction Correction to Density-Functional Approximations for many-electron Systems. Phys Rev B (1981) 23:5048–79. doi:10.1103/PhysRevB.23.5048
35. Burke K, Wagner LO. Dft in a Nutshell. Int J Quan Chem. (2013) 113:96–101. doi:10.1002/qua.24259
36. Hampel A, Beck S, Ederer C. Effect of Charge Self-Consistency in DFT+DMFT Calculations for Complex Transition Metal Oxides. Phys Rev Res (2020) 2:033088. doi:10.1103/PhysRevResearch.2.033088
37. Anisimov VI, Kondakov DE, Kozhevnikov AV, Nekrasov IA, Pchelkina ZV, Allen JW, et al. Full Orbital Calculation Scheme for Materials with Strongly Correlated Electrons. Phys Rev B (2005) 71:125119. doi:10.1103/physrevb.71.125119
38. Amadon B, Lechermann F, Georges A, Jollet F, Wehling TO, Lichtenstein AI. Plane-wave Based Electronic Structure Calculations for Correlated Materials Using Dynamical Mean-Field Theory and Projected Local Orbitals. Phys Rev B (2008) 77:205112. doi:10.1103/PhysRevB.77.205112
39. Aichhorn M, Pourovskii L, Vildosola V, Ferrero M, Parcollet O, Miyake T, et al. Dynamical Mean-Field Theory within an Augmented Plane-Wave Framework: Assessing Electronic Correlations in the Iron Pnictide Lafeaso. Phys Rev B (2009) 80:085101. doi:10.1103/PhysRevB.80.085101
40. Marzari N, Vanderbilt D. Maximally Localized Generalized Wannier Functions for Composite Energy Bands. Phys Rev B (1997) 56:12847–65. doi:10.1103/PhysRevB.56.12847
41. Wang R, Lazar EA, Park H, Millis AJ, Marianetti CA. Selectively Localized Wannier Functions. Phys Rev B (2014) 90:165125. doi:10.1103/PhysRevB.90.165125
42. Marzari N, Mostofi AA, Yates JR, Souza I, Vanderbilt D. Maximally Localized Wannier Functions: Theory and Applications. Rev Mod Phys (2012) 84:1419–75. doi:10.1103/RevModPhys.84.1419
43. Souza I, Marzari N, Vanderbilt D. Maximally Localized Wannier Functions for Entangled Energy Bands. Phys Rev B (2001) 65:035109. doi:10.1103/PhysRevB.65.035109
44. Karp J, Hampel A, Millis AJ. Dependence of DFT+DMFT Results on the Construction of the Correlated Orbitals. Phys Rev B (2021) 103:195101. doi:10.1103/PhysRevB.103.195101
45. Haule K. Exact Double Counting in Combining the Dynamical Mean Field Theory and the Density Functional Theory. Phys Rev Lett (2015) 115:196403. doi:10.1103/PhysRevLett.115.196403
46. Beck S, Hampel A, Parcollet O, Ederer C, Georges A. Charge Self-Consistent Electronic Structure Calculations with Dynamical Mean-Field Theory Using Quantum Espresso, Wannier90 and Triqs. arXiv (2021) 2111–10289.
47. Park H, Millis AJ, Marianetti CA. Density Functional versus Spin-Density Functional and the Choice of Correlated Subspace in Multivariable Effective Action Theories of Electronic Structure. Phys Rev B (2015) 92:035146. doi:10.1103/physrevb.92.035146
48. Pavarini E, Koch E, Lichtenstein A, Vollhardt DE. The LDA+DMFT Approach to Strongly Correlated Materials. Schriften des Forschungszentrums Jülich : Model Simulation (2011) 1. Record converted from VDB: 12.11.2012.
49. Slater LJ. Confluent Hypergeometric Functions. Cambridge, United Kingdom: Cambridge Univ. Press (1960).
50. Vaugier L, Jiang H, Biermann S. HubbardUand Hund exchangeJin Transition Metal Oxides: Screening versus Localization Trends from Constrained Random Phase Approximation. Phys Rev B (2012) 86:165105. doi:10.1103/PhysRevB.86.165105
51. Castellani C, Natoli CR, Ranninger J. Magnetic Structure ofV2O3in the Insulating Phase. Phys Rev B (1978) 18:4945–66. doi:10.1103/PhysRevB.18.4945
52. Kanamori J. Electron Correlation and Ferromagnetism of Transition Metals. Prog Theor Phys (1963) 30:275–89. doi:10.1143/PTP.30.275
53. Anisimov VI, Gunnarsson O. Density-functional Calculation of Effective Coulomb Interactions in Metals. Phys Rev B (1991) 43:7570–4. doi:10.1103/PhysRevB.43.7570
54. Aryasetiawan F, Imada M, Georges A, Kotliar G, Biermann S, Lichtenstein AI. Frequency-dependent Local Interactions and Low-Energy Effective Models from Electronic Structure Calculations. Phys Rev B (2004) 70:195104. doi:10.1103/physrevb.70.195104
55. Aryasetiawan F, Miyake T, Sakuma R. The LDA+DMFT approach to strongly correlated materials (Schriften des Forschungszentrums Jülich), chap. The Constrained RPA Method for Calculating the Hubard U from First-Principles (2011). p. 71–726.
57. Vogel D, Krüger P, Pollmann J. Self-interaction and Relaxation-Corrected Pseudopotentials for II-VI Semiconductors. Phys Rev B (1996) 54:5495–511. doi:10.1103/physrevb.54.5495
58. Filippetti A, Spaldin NA. Self-interaction-corrected Pseudopotential Scheme for Magnetic and Strongly-Correlated Systems. Phys Rev B (2003) 67:125109. doi:10.1103/PhysRevB.67.125109
59. Puphal P, Wu M, Fürsich K, Lee H, Pakdaman M, Bruin JAN, et al. Synthesis and Characterization of Ca-Substituted Infinite-Layer Nickelate 2121 Crystals. Sci Adv (2021) 7:eabl8091.
60. Lechermann F, Körner W, Urban DF, Elsässer C. Interplay of Charge-Transfer and mott-hubbard Physics Approached by an Efficient Combination of Self-Interaction Correction and Dynamical Mean-Field Theory. Phys Rev B (2019) 100:115125. doi:10.1103/physrevb.100.115125
61. Wang BX, Zheng H, Krivyakina E, Chmaissem O, Lopes PP, Lynn JW, et al. Synthesis and Characterization of Bulk Nd1−xSrxNiO2 and Nd1−xSrxNiO3. Phys Rev Mater (2020) 4:084409. doi:10.1103/physrevmaterials.4.084409
62. Si L, Xiao W, Kaufmann J, Tomczak JM, Lu Y, Zhong Z, et al. Topotactic Hydrogen in Nickelate Superconductors and Akin Infinite-Layer Oxides ABO2. Phys Rev Lett (2020) 124:166402. doi:10.1103/physrevlett.124.166402
63. Geisler B, Pentcheva R. Fundamental Difference in the Electronic Reconstruction of Infinite-Layer versus Perovskite Neodymium Nickelate Films on SrTiO3 (001). Phys Rev B (2020) 102:020502. doi:10.1103/physrevb.102.020502
64. Sakakibara H, Usui H, Suzuki K, Kotani T, Aoki H, Kuroki K. Model Construction and a Possibility of Cupratelike Pairing in a New D9 Nickelate Superconductor (Nd,Sr)NiO2. Phys Rev Lett (2020) 125:077003. doi:10.1103/PhysRevLett.125.077003
65. He R, Jiang P, Lu Y, Song Y, Chen M, Jin M, et al. Polarity-induced Electronic and Atomic Reconstruction at NdNiO2/SrTiO3 Interfaces. Phys Rev B (2020) 102:035118. doi:10.1103/PhysRevB.102.035118
66. Zhang H, Jin L, Wang S, Xi B, Shi X, Ye F, et al. Effective Hamiltonian for Nickelate Oxides Nd1−xSrxNiO2. Phys Rev Res (2020) 2:013214. doi:10.1103/PhysRevResearch.2.013214
67. Zhang J, Zheng H, Chen Y-S, Ren Y, Yonemura M, Huq A, et al. High Oxygen Pressure Floating Zone Growth and crystal Structure of the Metallic Nickelates R4Ni3O10 ( R=La,Pr ). Phys Rev Mater (2020) 4:083402. doi:10.1103/PhysRevMaterials.4.083402
68. Bernardini F, Olevano V, Blase X, Cano A. Infinite-layer Fluoro-Nickelates as D 9 Model Materials. J Phys Mater (2020) 3:035003. doi:10.1088/2515-7639/ab885d
69. Bernardini F, Cano A. Stability and Electronic Properties of LaNiO2/SrTiO3 Heterostructures. J Phys Mater (2020) 3:03LT01. doi:10.1088/2515-7639/ab9d0f
70. Liu Z, Xu C, Cao C, Zhu W, Wang ZF, Yang J. Doping Dependence of Electronic Structure of Infinite-Layer NdNiO2. Phys Rev B (2021) 103:045103. doi:10.1103/PhysRevB.103.045103
71. Plienbumrung T, Daghofer M, Oleś AM. Interplay between Zhang-Rice Singlets and High-Spin States in a Model for Doped NiO2 Planes. Phys Rev B (2021) 103:104513. doi:10.1103/PhysRevB.103.104513
72. Malyi OI, Varignon J, Zunger A. Bulk NdNiO2 Is Thermodynamically Unstable with Respect to Decomposition while Hydrogenation Reduces the Instability and Transforms it from Metal to Insulator. arXiv (2021) 2107–01790.
73. Peng C, Jiang HC, Moritz B, Devereaux TP, Jia C. Superconductivity in a Minimal Two-Band Model for Infinite-Layer Nickelates. arXiv (2021) 2110.07593. (2021).
74. Choubey P, Eremin IM. Electronic Theory for Scanning Tunneling Microscopy Spectra in Infinite-Layer Nickelate Superconductors. Phys Rev B (2021) 104:144504. doi:10.1103/PhysRevB.104.144504
75. Sawatzky GA. Superconductivity Seen in a Non-magnetic Nickel Oxide. Nature (2019) 572:592–3. doi:10.1038/d41586-019-02518-3
76. Nomura Y, Nomoto T, Hirayama M, Arita R. Magnetic Exchange Coupling in Cuprate-Analog D9 Nickelates. Phys Rev Res (2020) 2:043144. doi:10.1103/PhysRevResearch.2.043144
77. Hirayama M, Tadano T, Nomura Y, Arita R. Materials Design of Dynamically Stable D9 Layered Nickelates. Phys Rev B (2020) 101:075107. doi:10.1103/PhysRevB.101.075107
78. Jin H-S, Pickett WE, Lee K-W. Proposed Ordering of Textured Spin Singlets in a Bulk Infinite-Layer Nickelate. Phys Rev Res (2020) 2:033197. doi:10.1103/PhysRevResearch.2.033197
79. Petocchi F, Christiansson V, Nilsson F, Aryasetiawan F, Werner P. Normal State of Nd1−xSrxNiO2 from Self-Consistent GW+EDMFT. Phys Rev X (2020) 10:041047. doi:10.1103/PhysRevX.10.041047
80. Higashi K, Winder M, Kuneš J, Hariki A. Core-Level X-Ray Spectroscopy of Infinite-Layer Nickelate: LDA+DMFT Study. Phys Rev X (2021) 11:041009. doi:10.1103/PhysRevX.11.041009
81. Leonov I. Effect of Lattice Strain on the Electronic Structure and Magnetic Correlations in Infinite-Layer (Nd,Sr)NiO2. J Alloys Compounds (2021) 883:160888. doi:10.1016/J.JALLCOM.2021.160888
82. Lin H, Gawryluk DJ, Klein YM, Huangfu S, Pomjakushina E, von Rohr F, et al. Universal Spin-Glass Behaviour in Bulk LaNiO2, PrNiO2 and NdNiO2. arXiv (2021) 2104–14324.
83. Lee K-W, Pickett WE. Infinite-layerLaNiO2: Ni1+is notCu2+. Phys Rev B (2004) 70:165109. doi:10.1103/PhysRevB.70.165109
84. Choi M-Y, Pickett WE, Lee K-W. Fluctuation-frustrated Flat Band Instabilities in NdNiO2. Phys Rev Res (2020) 2:033445. doi:10.1103/PhysRevResearch.2.033445
85. Choi M-Y, Lee K-W, Pickett WE. Role of 4f States in Infinite-Layer NdNiO2. Phys Rev B (2020) 101:020503. doi:10.1103/PhysRevB.101.020503
86. Zeng S, Tang CS, Yin X, Li C, Li M, Huang Z, et al. Phase Diagram and Superconducting Dome of Infinite-Layer Nd1−xSrxNiO2 Thin Films. Phys Rev Lett (2020) 125:147003. doi:10.1103/PhysRevLett.125.147003
87. Li D, Wang BY, Lee K, Harvey SP, Osada M, Goodge BH, et al. Superconducting Dome in Nd1−xSrxNiO2 Infinite Layer Films. Phys Rev Lett (2020) 125:027001. doi:10.1103/PhysRevLett.125.027001
88. Gu Q, Li Y, Wan S, Li H, Guo W, Yang H, et al. Single Particle Tunneling Spectrum of Superconducting Nd1-xSrxNiO2 Thin Films. Nat Commun (2020) 11. doi:10.1038/s41467-020-19908-1
89. Goodge BH, Li D, Lee K, Osada M, Wang BY, Sawatzky GA, et al. Doping Evolution of the Mott-Hubbard Landscape in Infinite-Layer Nickelates. Proc Natl Acad Sci USA (2021) 118:e2007683118. doi:10.1073/pnas.2007683118
90. Wang BY, Li D, Goodge BH, Lee K, Osada M, Harvey SP, et al. Isotropic Pauli-Limited Superconductivity in the Infinite-Layer Nickelate Nd0.775Sr0.225NiO2. Nat Phys (2021) 17:473–7. doi:10.1038/s41567-020-01128-5
91. Zhao D, Zhou YB, Fu Y, Wang L, Zhou XF, Cheng H, et al. Intrinsic Spin Susceptibility and Pseudogaplike Behavior in Infinite-Layer LaNiO2. Phys Rev Lett (2021) 126:197001. doi:10.1103/PhysRevLett.126.197001
92. Lu H, Rossi M, Nag A, Osada M, Li DF, Lee K, et al. Magnetic Excitations in Infinite-Layer Nickelates. Science (2021) 373:213–6. doi:10.1126/science.abd7726
93. Osada M, Wang BY, Goodge BH, Lee K, Yoon H, Sakuma K, et al. A Superconducting Praseodymium Nickelate with Infinite Layer Structure. Nano Lett (2020) 20:5735–40. doi:10.1021/acs.nanolett.0c01392
94. Osada M, Wang BY, Lee K, Li D, Hwang HY. Phase Diagram of Infinite Layer Praseodymium Nickelate Pr1−xSrxNiO2 Thin Films. Phys Rev Mater (2020) 4:121801. doi:10.1103/PhysRevMaterials.4.121801
95. Osada M, Wang BY, Goodge BH, Harvey SP, Lee K, Li D, et al. Nickelate Superconductivity without Rare‐Earth Magnetism: (La,Sr)NiO 2. Adv Mater (2021) 33:2104083. doi:10.1002/adma.202104083
96. Ren X, Gao Q, Zhao Y, Luo H, Zhou X, Zhu Z. Superconductivity in Infinite-Layer Pr0.8Sr0.2NiO2 Films on Different Substrates. arXiv (2021) 2109.05761. (2021).
97. Zeng SW, Li CJ, Chow LE, Cao Y, Zhang ZT, Tang CS, et al. Superconductivity in Infinite-Layer Lanthanide Nickelates. arXiv (2021) 2105.13492. (2021).
98. Puphal P, Wu YM, Fürsich K, Lee H, Pakdaman M, Bruin JAN, et al. Synthesis and Characterization of Ca-Substituted Infinite-Layer Nickelate Crystals. arXiv (2021) 2106–13171.
99. Liu Z, Ren Z, Zhu W, Wang Z, Yang J. Electronic and Magnetic Structure of Infinite-Layer NdNiO2: Trace of Antiferromagnetic Metal. Npj Quan Mater. (2020) 5. doi:10.1038/s41535-020-0229-1
100. Jiang P, Si L, Liao Z, Zhong Z. Electronic Structure of Rare-Earth Infinite-Layer RNiO2(R=La,Nd). Phys Rev B (2019) 100:201106. doi:10.1103/PhysRevB.100.201106
101. Ryee S, Yoon H, Kim TJ, Jeong MY, Han MJ. Induced Magnetic Two-Dimensionality by Hole Doping in the Superconducting Infinite-Layer Nickelate Nd1−xSrxNiO2. Phys Rev B (2020) 101:064513. doi:10.1103/physrevb.101.064513
102. Zhang G-M, Yang Y-f., Zhang F-C. Self-doped Mott Insulator for Parent Compounds of Nickelate Superconductors. Phys Rev B (2020) 101:020501. doi:10.1103/PhysRevB.101.020501
103. Yang Y, Zhang GM. Self-doping and the Mott-Kondo Scenario for Infinite-Layer Nickelate Superconductors. arXiv (2021) 2110.11089. (2021).
104. Xia C, Wu J, Chen Y, Chen H. Dynamical Structural Instability and a New crystal-electronic Structure of Infinite-Layer Nickelates. arXiv (2021) 2110–12405.
105. Zaanen J, Sawatzky GA, Allen JW. Band Gaps and Electronic Structure of Transition-Metal Compounds. Phys Rev Lett (1985) 55:418–21. doi:10.1103/physrevlett.55.418
106. Lechermann F. Late Transition Metal Oxides with Infinite-Layer Structure: Nickelates versus Cuprates. Phys Rev B (2020) 101:081110. doi:10.1103/PhysRevB.101.081110
107. Shen Y, Sears J, Fabbris G, Li J, Pelliciari J, Jarrige I, et al. Role of Oxygen States in Square Planar D9−δ Nickelates arXiv:2110.08937. (2021).
108. Lechermann F. Doping-dependent Character and Possible Magnetic Ordering of NdNiO2. Phys Rev Mater (2021) 5:044803. doi:10.1103/physrevmaterials.5.044803
109. Stadler KM, Kotliar G, Weichselbaum A, von Delft J. Hundness versus Mottness in a Three-Band Hubbard-Hund Model: On the Origin of strong Correlations in Hund Metals. Ann Phys (2019) 405:365–409. doi:10.1016/j.aop.2018.10.017
111. Zhang R, Lane C, Singh B, Nokelainen J, Barbiellini B, Markiewicz RS, et al. Magnetic and F-Electron Effects in LaNiO2 and NdNiO2 Nickelates with Cuprate-like $$3{d}$3d_{x^{2}-Y^{2}}$fx170_{{x}^{2}-{y}^{2}}$$ Band Band. Commun Phys (2021) 4. doi:10.1038/s42005-021-00621-4
112. Katukuri VM, Bogdanov NA, Weser O, van den Brink J, Alavi A. Electronic Correlations and Magnetic Interactions in Infinite-Layer NdNiO2. Phys Rev B (2020) 102:241112. doi:10.1103/PhysRevB.102.241112
113. Wang Z, Zhang G-M, Yang Y-f., Zhang F-C. Distinct Pairing Symmetries of Superconductivity in Infinite-Layer Nickelates. Phys Rev B (2020) 102:220501. doi:10.1103/PhysRevB.102.220501
114. Adhikary P, Bandyopadhyay S, Das T, Dasgupta I, Saha-Dasgupta T. Orbital-selective Superconductivity in a Two-Band Model of Infinite-Layer Nickelates. Phys Rev B (2020) 102:100501. doi:10.1103/PhysRevB.102.100501
115. Momma K, Izumi F. VESTA 3for Three-Dimensional Visualization of crystal, Volumetric and Morphology Data. J Appl Cryst (2011) 44:1272–6. doi:10.1107/S0021889811038970
116. Lechermann F. From Basic Properties to the Mott Design of Correlated Delafossites. Npj Comput Mater (2021) 7:120. doi:10.1038/s41524-021-00586-6
Keywords: nickelates, correlated electron physics, downfolding, dynamical mean field theory, density functional theory, quantum embedding
Citation: Chen H, Hampel A, Karp J, Lechermann F and Millis AJ (2022) Dynamical Mean Field Studies of Infinite Layer Nickelates: Physics Results and Methodological Implications. Front. Phys. 10:835942. doi: 10.3389/fphy.2022.835942
Received: 15 December 2021; Accepted: 07 January 2022;
Published: 02 February 2022.
Edited by:
Antia Botana, Arizona State University, United StatesCopyright © 2022 Chen, Hampel, Karp, Lechermann and Millis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanghui Chen, hanghui.chen@nyu.edu