Integrative Analysis of lncRNA-mRNA Co-expression Provides Novel Insights Into the Regulation of Developmental Transitions in Female Varroa destructor
 Jiang-Li Wu
Jiang-Li Wu Ruo-Yang Hu
Ruo-Yang Hu Nan-Nan Li
Nan-Nan Li Jing Tan
Jing Tan Chun-Xue Zhou
Chun-Xue Zhou Bin Han
Bin Han Shu-Fa Xu
Shu-Fa Xu- 1Key Laboratory of Pollinating Insect Biology, Ministry of Agriculture and Rural Affairs, Institute of Apicultural Research, Chinese Academy of Agricultural Sciences, Beijing, China
- 2Department of Parasitology, Shandong University School of Basic Medicine, Jinan, China
Varroa destructor is a major pathogenic driver of the Western honeybee colony losses globally. Understanding the developmental regulation of V. destructor is critical to develop effective control measures. Development is a complex biological process regulated by numerous genes and long non-coding RNAs (lncRNAs); however, the underlying regulation of lncRNAs in the development of V. destructor remains unknown. In this study, we analyzed the RNA sequencing (RNA-Seq) data derived from the four stages of female V. destructor in the reproductive phase (i.e., egg, protonymph, deutonymph, and adult). The identified differentially expressed mRNAs and lncRNAs exhibited a stage-specific pattern during developmental transitions. Further functional enrichment established that fat digestion and absorption, ATP-binding cassette (ABC) transporters, mitogen-activated protein kinase (MAPK) signaling pathway, and ubiquitin-proteasome pathway play key roles in the maturation of female V. destructor. Moreover, the lncRNAs and mRNAs of some pivotal genes were significantly upregulated at the deutonymph stage, such as cuticle protein 65/6.4/63/38 and mucin 5AC, suggesting that deutonymph is the key stage of metamorphosis development and pathogen resistance acquisition for female V. destructor. Our study provides novel insights into a foundational understanding of V. destructor biology.
Introduction
Varroa destructor has been and still is the greatest biotic threat to the Western honeybee (Apis mellifera) worldwide since its migration from the Eastern honeybee (Apis cerana). As an obligate ectoparasitic mite, V. destructor feeds primarily on the fat body of brood and adult honeybees and thus results in decreased body weight, malformation, as well as a shorter life span of adult bees (Rosenkranz et al., 2010; Ramsey et al., 2019). V. destructor infestation is also intimately coupled to the pathogen transmission within and between honeybee colonies, which impairs immunity defenses at both the individual and colony levels. Great efforts have been made in developing solutions to efficiently control the mite so far; however, the combination of the mite with other biotic and abiotic stressors continues to shake the beekeeping and pollination industries (Noël et al., 2020).
Female V. destructor mites have two distinct stages in their life cycle, the dispersal phase on adult bees, and the reproductive phase, which takes place inside capped brood cells during bee development (Traynor et al., 2020). In the reproductive phase, the mother mite (foundress) completes reproduction by feeding on the fat body of the larva (Li et al., 2019). Depending on the chemical signal stimulus (kairomones), the foundress mites invade appropriately aged larval cells to oviposition, resulting in a single son (about 6.6 days) and up to 7–9 daughters (about 5.8 days) (Martin, 1994; Boecking and Genersch, 2008; Traynor et al., 2020). Once the eggs hatch, the juveniles develop from protonymphs to deutonymphs before molting into adults. Previous studies showed that the development of V. destructor is a complex process, in which many genes/proteins cooperate to regulate developmental transition and sexual differentiation (Cornman et al., 2010; McAfee et al., 2017; Mondet et al., 2018). It has also been shown that the development of V. destructor relies on effective coordination between physiological readiness and responsiveness to host cues (Evans and Cook, 2018). Although these advances have improved our understanding of this complex parasite, our knowledge of V. destructor biology is still insufficient, especially the regulation during the developmental transition from egg to adult remains to be explored.
Long non-coding RNAs (lncRNAs) are an important class of pervasive genes with more than 200 bases that function as regulators with less or no protein-coding capacity (Wang and Chang, 2011; Quinn and Chang, 2016). LncRNAs have been shown to play vital roles in regulating gene expression at transcription, RNA processing, and translation levels through cis (near the site of lncRNA production) or trans (co-expressed with their target gene) ways (Guttman et al., 2011; Batista and Chang, 2013; Li Y.R. et al., 2020). Growing studies suggest that lncRNAs participate in a broad range of biological processes (BPs). For example, in honeybees, lncRNAs have been engaged in the regulation of the oviposition and ovary size of queens (Humann et al., 2013; Chen et al., 2017; Chen and Shi, 2020), neural function (Sawata et al., 2004; Kiya et al., 2012), DNA methylation (Wedd et al., 2016), pathogen infection (Guo et al., 2018a,b), pesticide metabolism (Jayakodi et al., 2015; Chen et al., 2019; Fent et al., 2020), and behavioral transition (Liu et al., 2019). For V. destructor, however, the only report provided the first catalog of lncRNA profile in gravid female mites, suggesting that lncRNAs play a critical role in cellular and biological progresses (Lin et al., 2020). Nevertheless, so far, the potential biological function of lncRNAs during the developmental transitions of female V. destructor and their regulatory roles are still poorly understood.
To address these gaps in knowledge, in this study, we analyzed the expression profiles of lncRNAs and mRNAs across all the developmental stages of female V. destructor (i.e., egg, protonymph, deutonymph, and adult). Using RNA sequencing (RNA-Seq), we identified a large number of new lncRNAs and mRNAs, expanding the genetic database of V. destructor to a new depth. Furthermore, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of the differentially expressed target genes of lncRNAs and mRNAs revealed critical pathways that play key roles in the developmental regulation of V. destructor at different stages. Our findings thus indicate significant roles of lncRNAs in orchestrating the development of female V. destructor mites and are potentially useful to develop effective methods for controlling this extremely threatening parasite.
Materials and Methods
Samples Collection and Total RNA Extraction
A total of three A. mellifera honeybee hives with mite populations were used in this study. All colonies were bred at the apiary of the Institute of Apicultural Research, Chinese Academy of Agricultural Sciences, Beijing, under the same conditions. No acaricide treatment was applied in this study to avoid pesticide bias. According to the previous sample collection method (Dietemann et al., 2013; McAfee et al., 2017), V. destructor families were collected from a single A. mellifera colony. Eggs, protonymph, deutonymph, and adults were transferred directly to tubes using a soft paintbrush and soft tweezers. Approximately 200 individuals were pooled for each replicate (four developmental stages, n = 3 for each stage). All collected samples were immediately fresh-frozen using liquid nitrogen and stored at −80°C for further processing.
Total RNA of each sample was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, United States) according to the recommendations of the manufacturer. Extracted RNA was treated with Turbo DNA-Free Kit (Ambion Inc., Foster City, CA, United States) to remove contaminating genomic DNA. The quality and quantity of total RNA were evaluated using the A260:A280 ratio. NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, United States) was used to determine the concentration of RNA, and the integrity was analyzed with the Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, United States) using the Agilent RNA 6000 Nano Kit.
Complementary DNA Library Construction and Sequencing
First, before library construction, ribosomal RNA (rRNA) was removed using the Ribo-Zero™ rRNA Removal Kit (Illumina Inc., San Diego, CA, United States). Second, the generation of libraries for sequencing was made using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, United States) by following the instructions of the manufacturer. The products were then purified using a TruSeq RNASample Prep Kit v2 (NEB, Ipswich, MA, United States), and library concentration was measured using the Quant-iT PicoGreen Double-Stranded DNA Assay Kit (Invitrogen, Carlsbad, CA, United States). Fragment size was estimated using the Agilent High Sensitivity DNA Analysis Kit with the Agilent Bioanalyzer 2100 system and quantified by the quantitative real-time PCR (qRT-PCR) using the KAPA Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems, Wilmington, MA, United States). Finally, the libraries were sequenced on an Illumina HiSeq Xten platform. The sequencing data have been submitted to the NCBI Sequence Read Archive (SRA) and are accessible through the BioProject accession number: PRJNA777703.
Quality Analysis, Mapping, and Transcriptome Assembly
The raw reads were filtered using the SOAPnuke (v1.5.2, parameters: −l 15 −q 0.2 −n 0.05 −i), and clean reads were obtained by removing contaminating adapter molecules, reads containing poly-N, and low-quality reads. All following analyses were performed based on the high-quality clean data. Sequenced reads in the FASTQ format were aligned to the V. destructor genome (Vdes_3.01) by using HISAT2 (v2.0.4, parameters: –phred64 –sensitive –no-discordant –no-mixed −l 1 −X 1000), and then the mapped reads were assembled using StringTie (v1.0.4, parameters: −f 0.3 −j 3 −c 5 −g 100 −s 10000 −p 8).
Coding Capacity Analysis
To identify new transcripts, all the reconstructed transcripts of each sample were aligned to the reference genome by using Cuffcompare; then, the novel transcripts were filtered and assembled to obtain putative transcripts. The putative novel lncRNAs were screened by the following steps: first, transcripts with transcript length >200 bp were selected; second, transcripts with Fragments Per Kilobase of exon per Million fragments mapped (FPKM)≥0.5 expression level were selected; third, transcripts with coverage >1 were selected; fourth, transcripts with one of the five class codes, namely, “i,” “j,” “u,” “o,” and “x,” were included (Sun et al., 2012), which were potentially recognized as novel ones. In addition, to further distinguish lncRNAs and mRNAs, the coding capacity of the novel transcripts was jointly calculated by Coding Potential Calculator (CPC), Coding-Non-Coding Index (CNCI), txCdsPredict, and Pfam-scan. Transcripts revealing a coding potential with CPC score < 0, CNCI score < 0, txCdsPredict score < 500, and Pfam-scan < 0.001 were identified as lncRNAs; others were considered as mRNAs. The transcript was identified as either lncRNA or mRNA only when the results were consistent among at least three judgment methods.
LncRNA and mRNA Expression Analyses
Gene expression was calculated using the normalized expression values as FPKM by RSEM (v1.2.12). Then, the differential expression analysis among the four groups was performed using the DEGseq software. Adjusted P-value ≤ 0.001 and absolute value of fold change ≥2 were set as the threshold for significantly differential expression of the coding genes and lncRNAs.
To predict the cis-target genes of identified lncRNAs, the adjacent protein-coding gene was searched within the range of 10-kb upstream or 20-kb downstream of a lncRNA (Yang et al., 2021). Then, the expressions of the lncRNAs and their candidate mRNAs were calculated by Spearman’s and Pearson’s correlation coefficients, and spearman_cor ≥ 0.6 and pearson_cor ≥ 0.6 were considered as the correlated expressions.
Functional Analyses
The differentially expressed mRNAs or lncRNA target genes were used for functional enrichment by the GO analysis and the KEGG pathway analysis. For the GO analysis, the differential expression genes (DEGs) were classified according to the principle classification of GO,2 and the number of differential transcripts included in each GO category was counted. For the KEGG3 enrichment analysis, the hypergeometric distribution test was used, and pathways with uncorrected P-value ≤ 0.05 were considered as significantly enriched.
Quantitative Real-Time PCR Validation
Three lncRNAs and their target mRNAs were selected for quantitative real-time PCR (qRT-PCR) to validate the RNA-Seq results. qRT-PCR was performed using SYBR Fast qPCR Kit Master Mix (2×) (KAPA Biosystems, Boston, MA, United States) on a QuantStudio 6&7 Flex Real-Time PCR system (Applied Biosystems, Foster City, CA, United States) according to the instructions of the manufacturer. All primers used in this study are listed in Supplementary Table 1. GAPDH was used as the reference gene. The conditions were set as follows: 95°C for 2 min, followed by 40 cycles (95°C for 3 s and 60°C for 30 s); then, 95°C for 15 s and 60°C for 15s; finally, 95°C for 15 s. The relative expression levels were then calculated based on the 2–ΔΔCT method.
Results
Overview of RNA-Sequencing
In this study, a total of 12 complementary DNA (cDNA) libraries were constructed and sequenced on the Illumine HiSeq Xten platform by using total RNA from each sample, and each sample was measured in three biological replicates (Supplementary Figure 1). After quality control, 12.64 Gb of clean reads were obtained, and the Q30 ranged from 83.54 to 97.66% for each sample. In total, 37.78–70.31% of clean reads from each library were mapped onto the V. destructor reference genome, and a total of 42,555 transcriptions were detected (Supplementary Table 2).
Identification and Characteristics of mRNAs and Long Non-coding RNAs
After mapping and coding capacity analysis, 9,760 (3,075 known and 6,685 novel) non-coding transcripts and 32,795 (28,091 known and 4,704 novel) protein-coding transcripts were obtained, which were used for downstream analyses. To comprehensively examine the differences between the two transcript species, we compared different characteristics (i.e., transcript number, transcript length, and exon number) of mRNAs and lncRNAs. The average length of mRNA was longer than that of lncRNA. Compared to mRNAs, a smaller number of exons and genes were identified in lncRNAs (Figure 1).
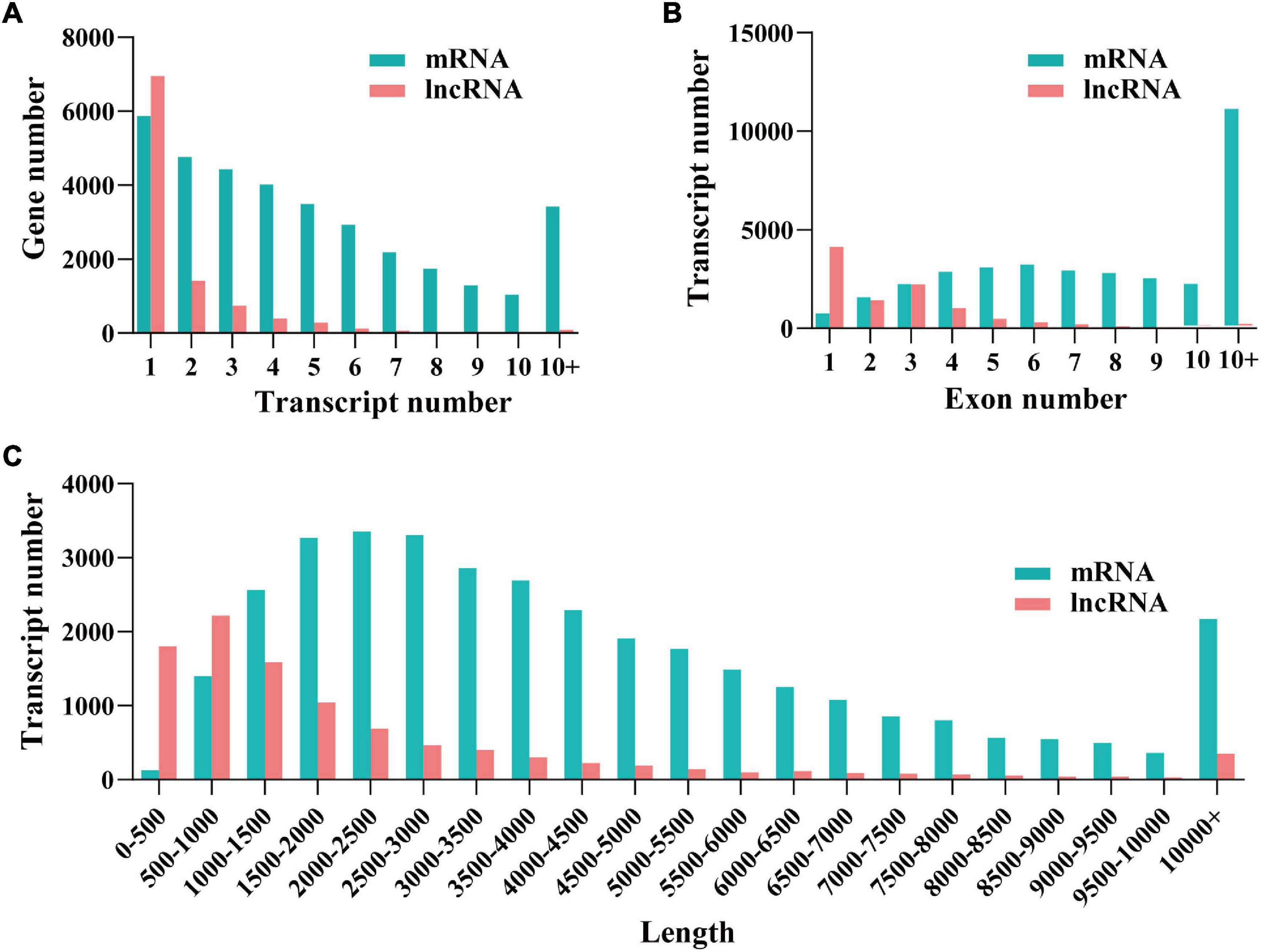
Figure 1. Identification and characterizationof mRNAs and long non-coding RNAs (lncRNAs). The distribution of transcript number (A), exon number (B), and transcript length (C) of lncRNAs and mRNAs.
Dynamic Expression Patterns of mRNAs and Long Non-coding RNAs
By calculating the gene expression levels from the RNA-Seq data using RSEM, 12,804 mRNAs and 7,954 lncRNAs were quantified in at least one biological replicate (FPKM > 0), and all lncRNAs tended to be expressed at a lower level than that of mRNAs (Figure 2A and Supplementary Tables 3, 4). For each stage, 11,750 mRNAs and 5,880 lncRNAs were identified in eggs, 11,502 mRNAs and 5,605 lncRNAs in protonymphs, 10,612 mRNAs and 3,572 lncRNAs in deutonymphs, and 11,241 mRNAs and 4,409 lncRNAs in adults (Supplementary Figure 2). Based on the expression changes, principal component analysis (PCA) was used to evaluate the distance relationship among the 12 samples. PCA score plot showed a clear separation of the four developmental stages (Figures 2B,C). Then, to understand the expression patterns of mRNA and lncRNA in mite samples, we generated the heatmaps using hierarchical clustering analyses to show the different changes of mRNAs and lncRNAs according to the development of female V. destructor(Figures 2D,E).
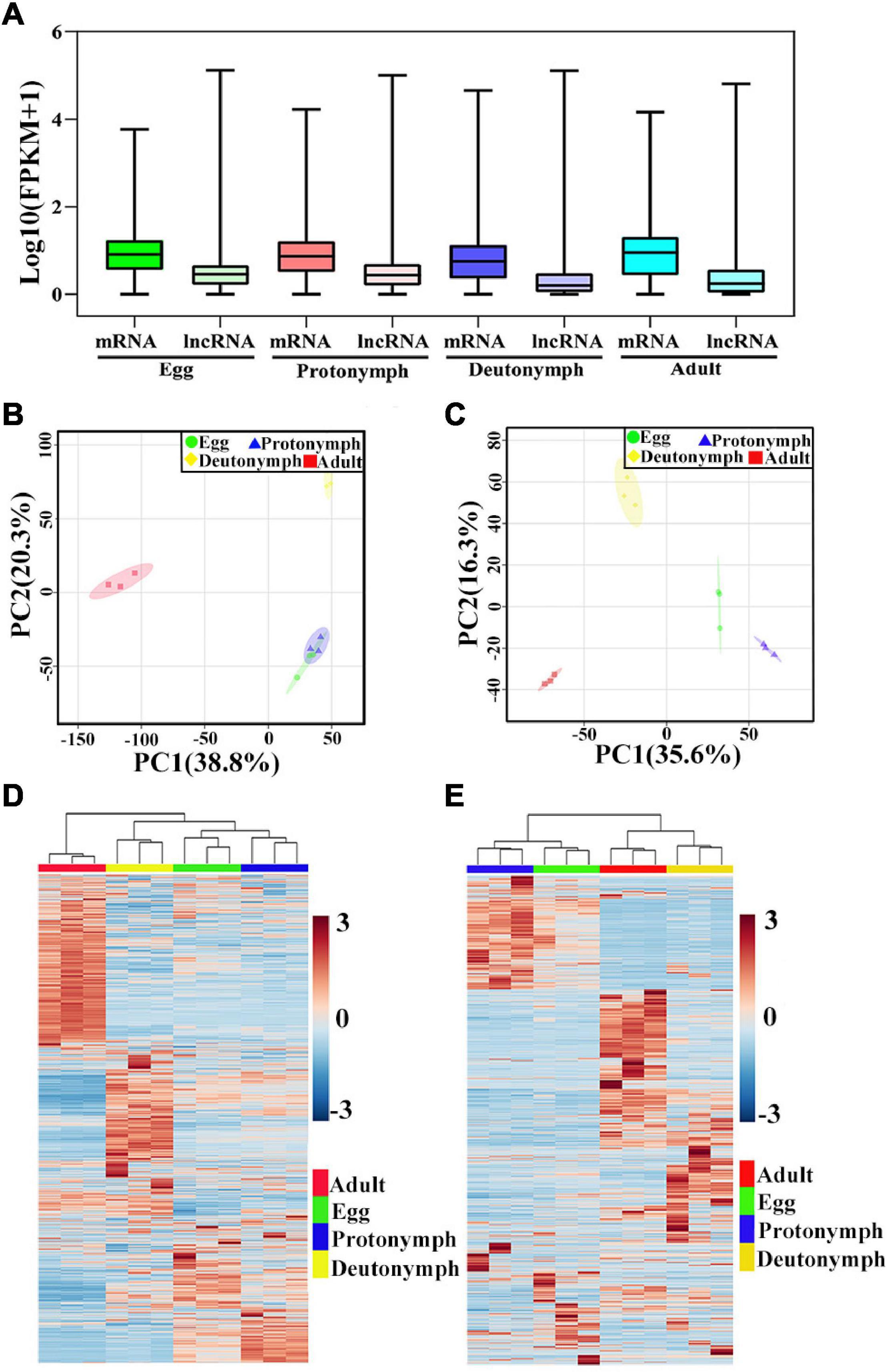
Figure 2. Expression profiles of mRNAs and long non-coding RNAs (lncRNAs). (A) The expression level of mRNAs and lncRNAs in four developmental stages. (B) Principal component analysis (PCA) based on mRNA. (C) PCA based on lncRNA. (D) Heatmap of mRNAs in the expression pattern profiles. (E) Heatmap of lncRNAs in the expression pattern profiles. Blue color indicates low expression, and red color denotes high expression.
To further investigate the expression pattern of mRNAs and lncRNAs during the four developmental stages of female V. destructor, the Short Time-series Expression Miner (STEM) was applied to cluster mRNAs and lncRNAs with similar expression patterns, respectively. A total of 12 models were established for mRNAs and lncRNAs, but only five and three were significantly enriched (Bonferroni-adjusted P-value ≤ 0.05), respectively (Figures 3A,B). Given that the mRNAs with similar expression patterns may also be functionally connected, we subsequently performed functional enrichment for each mRNA model and found intracellular membrane-bounded organelle ATP-binding cassette (ABC) transporters (GO: 0043231), regulation of protein serine/threonine kinase activity (GO: 0071900), the cytosolic ribosome (GO: 0022626), the primary metabolic process (GO: 0044238), and the ribonuclease T2 activity (GO: 0033897) as the leading terms in each model.
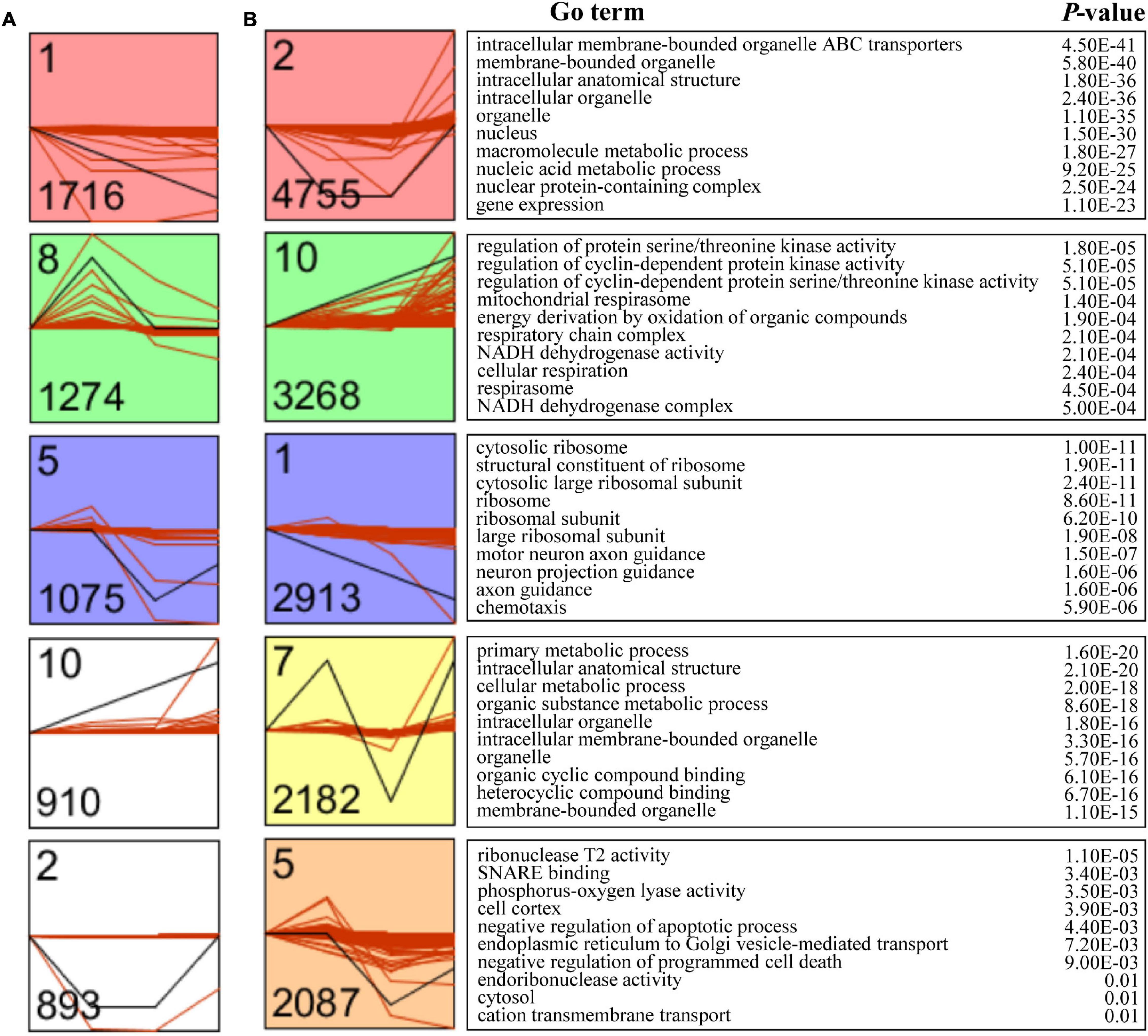
Figure 3. Time-series model of long non-coding RNAs (lncRNAs) and mRNAs.Time-series modules of lncRNAs (A) and mRNAs (B). Numbers in the top left corner indicate module number. Numbers in the lower left corner indicate the numbers of mRNAs or lncRNAs in each module. The Gene Ontology (GO) analysis of transcripts within clusters after the Fragments Per Kilobase of exon per Million fragments mapped (FPKM) filtering identified the associated enriched GO terms with corresponding enrichment P-value shown on right.
Differential Expression Analysis of mRNAs and Long Non-coding RNAs
To explore the key mRNAs and lncRNAs involved in the developmental regulation of female V. destructor, the data were pairwise compared within three groups (i.e., protonymph vs. egg, deutonymph vs. protonymph, and adult vs. deutonymph) (Supplementary Tables 5, 6). Consequently, in the protonymph vs. egg comparison, 1,085 differentially expressed mRNAs were detected, of which 498 were upregulated and 587 were downregulated (Supplementary Figure 3A). A total of 862 differentially expressed lncRNAs were detected, of which 454 were upregulated and 408 were downregulated (Figure 4A). In the deutonymph vs. protonymph comparison, 2,461 differentially expressed mRNAs were detected, of which 664 were upregulated and 1,797 were downregulated (Supplementary Figure 3B). A total of 1,963 differentially expressed lncRNAs were detected, of which 170 were upregulated and 1,793 were downregulated (Figure 4B). In the adult vs. deutonymph comparison, 5,429 differentially expressed mRNAs were detected, of which 3,992 were upregulated and 1,437 were downregulated (Supplementary Figure 3C). A total of 1,826 differentially expressed lncRNAs were detected, of which 1,112 were upregulated and 714 were downregulated (Figure 4C).
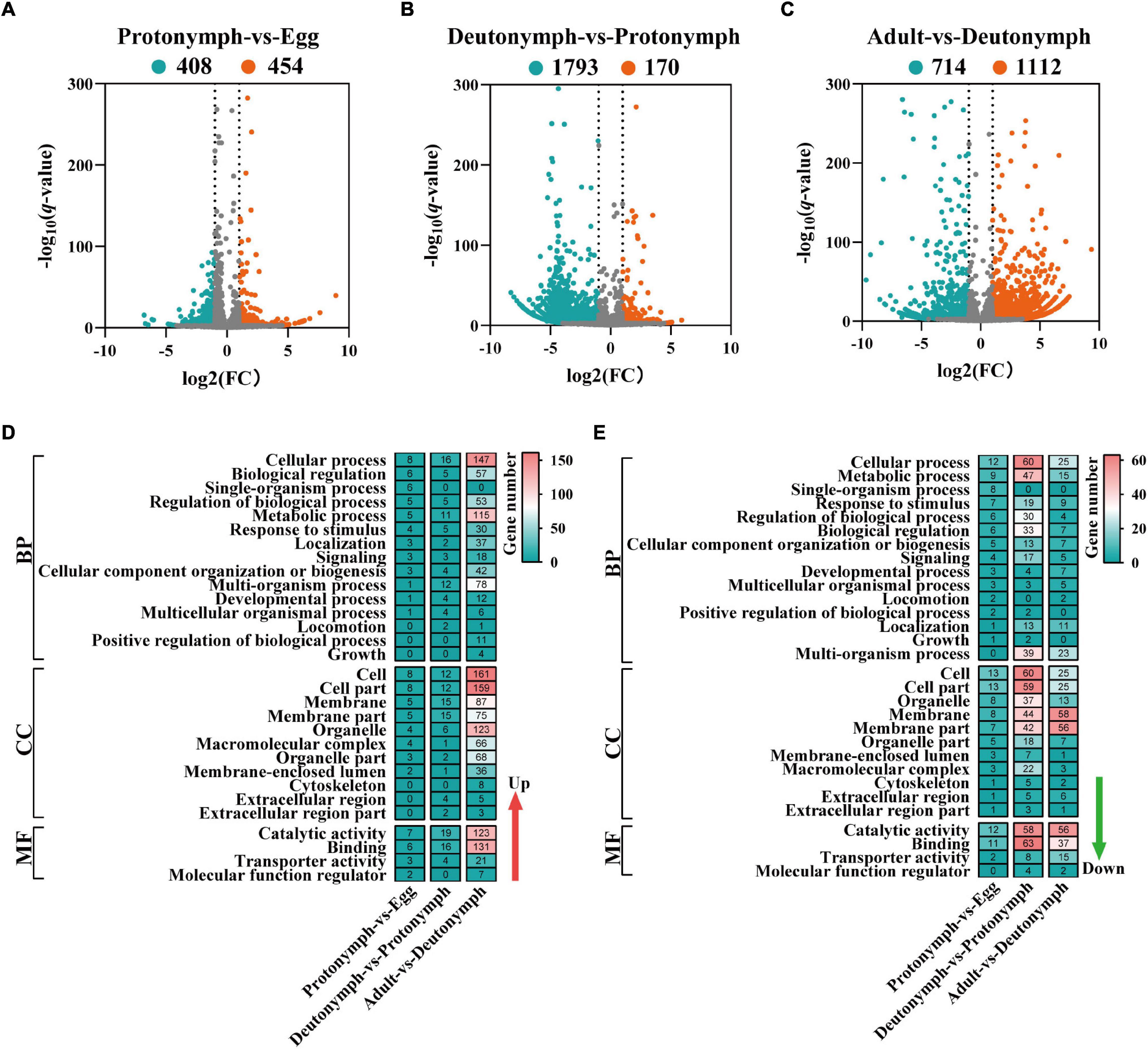
Figure 4. Differentially expressed long non-coding RNAs (lncRNAs) and functional analysis in differently compared libraries. (A) Volcano plot of the lncRNAs between protonymph and egg. (B) Volcano plot of the lncRNAs between deutonymph and protonymph. (C) Volcano plot of the lncRNAs between adult and deutonymph. The left blue points represent significantly decreased lncRNAs; gray points represent lncRNAs without significant changes; the right red points represent significantly increased lncRNAs. (D) The Gene Ontology (GO) analysis of the upregulated differentially expressed lncRNAs. (E) The GO analysis of the downregulated differentially expressed lncRNAs.
Gene Ontology Functional Analysis of mRNAs and Long Non-coding RNAs
To address how lncRNAs interact with their target genes (mRNAs) to regulate the developmental process of V. destructor, the potential targets of lncRNAs by cis-regulation were predicted. A total of 4,089 target genes within the range of 10-kb upstream or 20-kb downstream were predicted in the cis role (Supplementary Table 7). To better understand the gene regulation involved in the developmental transitions of female V. destructor, especially at the BP, the molecular function (MF), and the cellular component (CC) levels, we clustered the GO annotation of the differentially expressed mRNAs (Supplementary Figures 3D,E) and lncRNAs (Figures 4D,E). Based on the BP in the GO classification, the differentially expressed mRNAs and lncRNAs were classified into different functional categories, and the top five GO terms were enriched in “cellular process,” “metabolic process,” “single-organism process,” “biological regulation,” and “regulation of biological process.” In the CC domain, the top five GO terms were “cell,” “cell part,” “membrane,” “membrane part,” and “organelle.” In the MF, “catalytic activity,” “binding,” “transporter activity,” and “signal transducer activity” were the most highly represented terms (Supplementary Tables 8, 9).
Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of mRNAs and Long Non-coding RNAs
To gain further understanding of the biological functions of the differentially expressed lncRNAs and mRNAs during developmental transitions, the KEGG enrichment analyses of the mRNAs and lncRNA target genes in the three comparison groups were performed (Supplementary Tables 10, 11). In this study, we only selected the significantly enriched terms in the KEGG pathway analysis for each comparison, and the associations with human disease pathways were excluded. For protonymph vs. egg, deutonymph vs. protonymph, and adult vs. deutonymph comparisons, 16, 31, and 25 pathways that were highly enriched with mRNAs were detected, respectively. “ABC transporters” and “Hippo signaling pathway” were significantly enriched in the protonymph vs. egg comparison, “Glycosylphosphatidylinositol (GPI)-anchor biosynthesis” and “Thiamine metabolism” were significantly enriched in the deutonymph vs. protonymph comparison, and “Ribosome biogenesis in eukaryotes” and “RNA polymerase” were significantly enriched in the adult vs. deutonymph comparison (Supplementary Figure 4).
Notably, 5, 37, and 15 pathways were significantly enriched in the three comparison groups (i.e., protonymph vs. egg, deutonymph vs. protonymph, and adult vs. deutonymph), respectively. For example, “Fat digestion and absorption” and “ABC transporters” were significantly enriched in the protonymph vs. egg comparison; “Valine, leucine, and isoleucine degradation” and “D-Glutamine and D-glutamate metabolism” were significantly enriched in the deutonymph vs. protonymph comparison; and “Fat digestion and absorption” and “Riboflavin metabolism” were significantly enriched in the adult vs. deutonymph comparison (Figure 5).
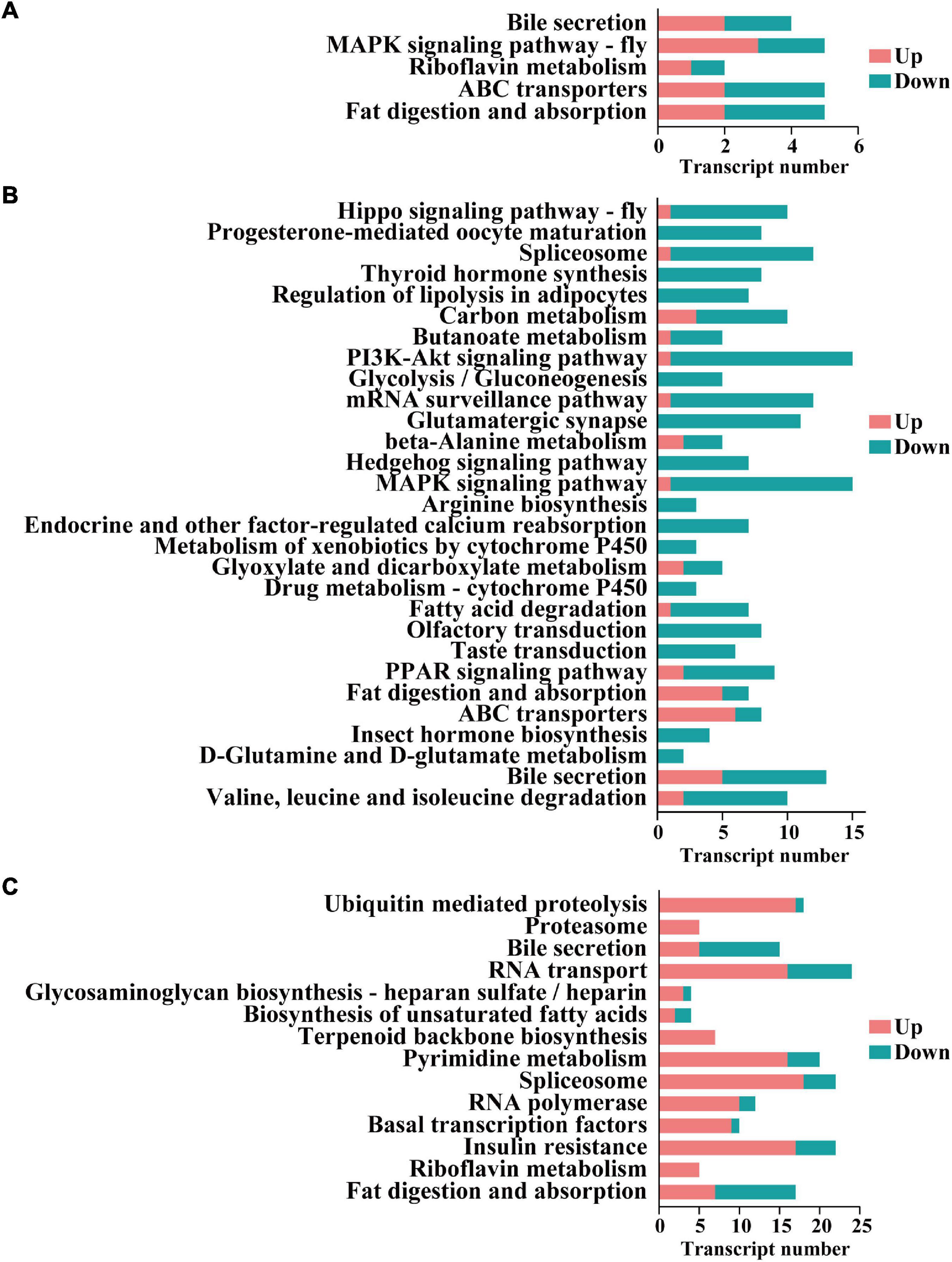
Figure 5. The enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the differentially expressed lncRNA. (A) Cis-target genes in the protonymph vs. egg comparison group. (B) Cis-target genes in the deutonymph vs. protonymph comparison group. (C) Cis-target genes in the adult vs. deutonymph comparison group.
Validation of mRNA and lncRNA
To verify the RNA-Seq results, three predicted lncRNAs (i.e., XR_002672204.1, LTCONS_00014321, and LTCONS_00011419) and their target genes (i.e., cuticle protein 65, ABCG5, and MUC5AC) were selected, and their expression levels were detected by qRT-PCR. As shown in Figure 6, the expression of lncRNA XR_002672204.1-cuticle protein 65, LTCONS_00014321-ABCG5, and LTCONS_00011419-MUC5AC was high in the deutonymph stage compared to the other stages. Moreover, all three lncRNAs and their target genes showed similar expression patterns.
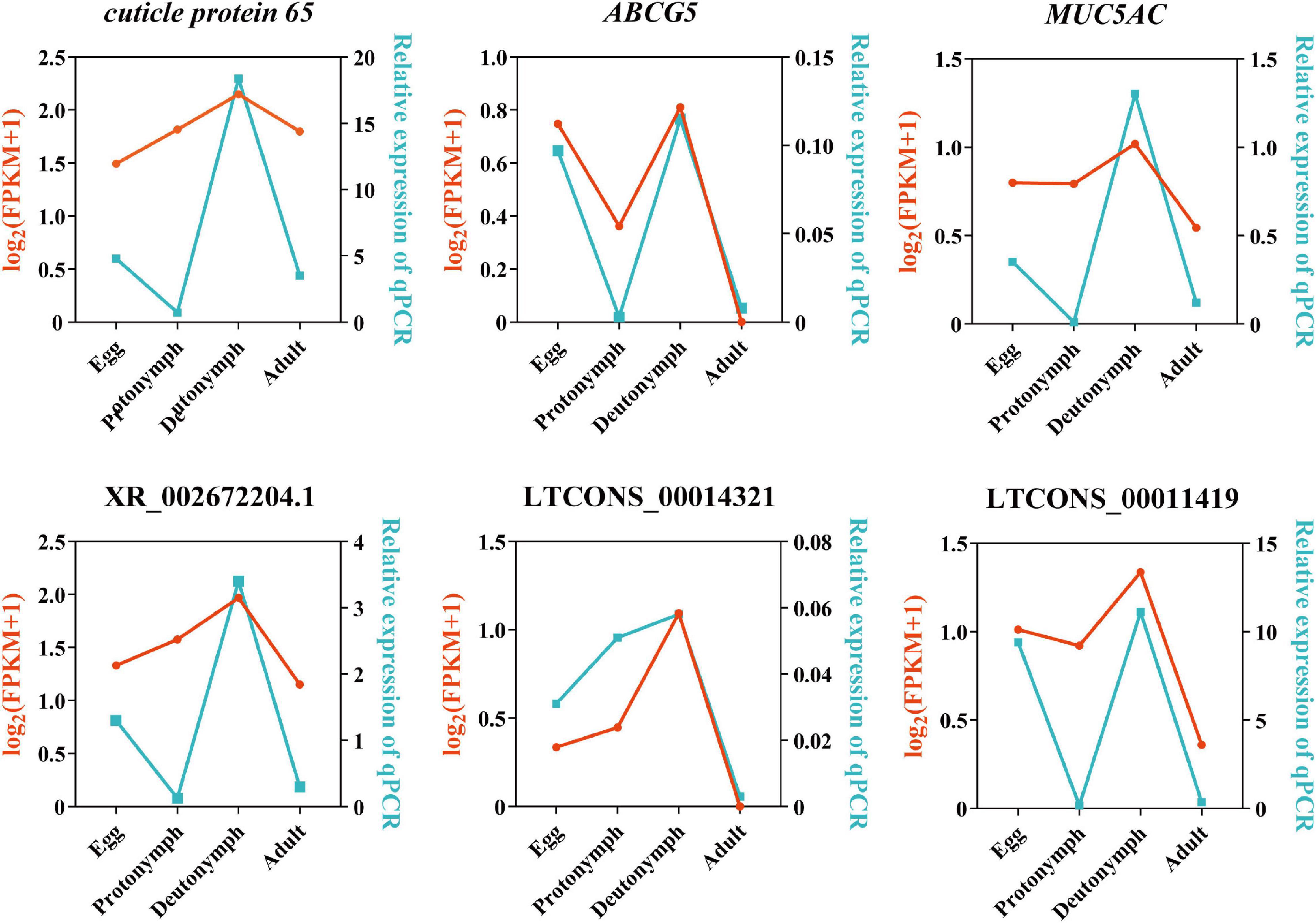
Figure 6. Validation of three significantly differentially expressed long non-coding RNAs (lncRNAs) and their target mRNAs by quantitative PCR (qPCR). X-axis represents the four developmental stages. Y-axis indicates the relative expression of each gene; red lines are log2(FPKM + 1) values of RNA-Seq, and the blue lines show relative expression by qPCR.
Discussion
The parasite V. destructor is an obligate ectoparasite and a highly invasive pest to honeybees (Nazzi and Le Conte, 2016). Due to the limited history of host-parasite coevolution, A. mellifera is suffering from V. destructor infestation. The specific parasitic life and increasing resistance to acaricides make developing efficient treatment strategies for V. destructor even more challenging. To gain a better understanding of the developmental regulation of female V. destructor, in this study, we performed an integrative analysis of lncRNA-mRNA co-expression crossing all the developmental stages (i.e., egg, protonymph, deutonymph, and adult). Consequently, the quantification analysis found 1,085, 2,461, and 5,429 mRNAs, and 862, 1,963, and 1,826 lncRNAs were differentially expressed in pairwise comparison (protonymph vs. egg, deutonymph vs. protonymph, and adult vs. deutonymph). Further functional enrichment revealed that fat digestion and absorption, ABC transporters, mitogen-activated protein kinase (MAPK) signaling pathway, and ubiquitin-proteasome pathway play key roles in the developmental transitions of female V. destructor.
Females of V. destructor show a development-specific pattern of lncRNA and mRNA expression. LncRNAs are an essential transcriptome component involved in the gene expression regulation in nearly all organisms studied (Fatica and Bozzoni, 2014; Gardini and Shiekhattar, 2015; Jarroux et al., 2017). In accordance with the lncRNA screening results of some extensively studied species, such as A. mellifera, Panonychus citri, and Tetranychus cinnabarinus (Zhang et al., 2014; Kim et al., 2018; Chen et al., 2019; Feng et al., 2020; Li Y. et al., 2021), our data showed relatively short length, low exon numbers, low transcript numbers, and generally low expression levels of V. destructor lncRNAs, implying that these features of lncRNAs were conserved in V. destructor. Accumulating evidence suggests that the expression profiles of lncRNAs exhibit tissue-, stage-, oocyte-, and/or development-specific patterns across species (Necsulea et al., 2014; Schor et al., 2018; Wang et al., 2020). In this study, we found that the number of identified mRNAs and lncRNAs gradually decreased from egg stage to deutonymph stage and then increased in the adult stage; however, the identified egg-specific and adult-specific lncRNAs and mRNAs were more than the other two stages, thus suggesting that gene expressions are more actively regulated in egg and adult stages. Furthermore, our quantitative results showed that, in the transition from protonymph to adult, the number of upregulated lncRNAs and mRNAs were gradually increased, indicating that more genes were regulated during developmental maturation.
Specific functional pathways are consolidated by lncRNA regulation at different developmental stages of female V. destructor. LncRNAs play regulatory roles in a wide spectrum of BPs, such as cell proliferation, cell differentiation and development, cell cycle, metabolism, apoptosis, and immune response (Wang et al., 2011; Batista and Chang, 2013; Geisler and Coller, 2013; Delas and Hannon, 2017). Particularly, in insects, lncRNAs have been implicated in mediating developmental processes and behavioral changes (Liu et al., 2019; Li G. et al., 2021; Yang et al., 2021). Our KEGG analysis of upregulated lncRNAs found that the fat digestion and absorption pathway was enriched in all the developmental stages of female V. destructor, which corresponds to the fact that lipid metabolism is fundamentally important to meet the energetic demands in growth and reproduction in insects (Arrese and Soulages, 2010). However, our results showed that energy was generated and utilized diversely at different developmental stages of V. destructor. During embryogenesis, energy was released by lipid mobilization to invest in the formation and development of organs; while, nymph and adult feed directly on honeybee fat bodies to obtain energy for development and reproduction (Zalewski et al., 2016; Ramsey et al., 2019). Noteworthily, in the fat digestion and absorption pathway, more upregulated lncRNAs were identified in deutonymph mites, echoing the notion that the deutonymphs have large energetic requirements to support energy-intensive developmental processes, such as metamorphosis (McAfee et al., 2017). In concert with this, ABC transporters and the Hippo signaling pathway were also found mainly elevated in deutonymphs. ABC transporters have been associated with the mite molting process (Li G. et al., 2021), with ABCGs particularly involved in pesticide transport and resistance (Dermauw and Van Leeuwen, 2014; Gott et al., 2017; Li Z. et al., 2020). We found that lncRNA LTCONS_00014321 and its target genes ABCG5 and ABCG8 were significantly upregulated in deutonymphs, implying that detoxification, metabolism, and especially metamorphosis are prominently reinforced in the deutonymph stage. Similarly, the upregulated Hippo signaling pathway is supposed to boost cell proliferation, tissue growth and differentiation, and organ migration in the deutonymph stage (Meng et al., 2016). In contrast, MAPK signaling was found consolidated in protonymphs. Given the functions of MAPK signaling in governing innate immune responses of mite (Tian et al., 2020), the upregulated lncRNAs and their target genes in this pathway might be correlated with the initial responses of V. destructor to environmental stressors such as pesticides (Lu et al., 2017). V. destructor is a well-known vector for the spreading of different honeybee viruses (Evans and Cook, 2018; Traynor et al., 2020). The ubiquitin-proteasome pathway of the host is usually employed or manipulated by viruses for their own needs (Gao and Luo, 2006); therefore, our results that the upregulated lncRNAs were mainly enriched in the adult stage may reflect the active promotion of virus-vector interactions (Gupta et al., 2019).
More evidence suggests that the deutonymph stage is crucial during the development of female V. destructor. Cuticle development is a fundamental physiological process in insect molting, keeping pace with growth, and metamorphosis (Liu et al., 2020; Li G. et al., 2021). During the maturation of V. destructor, nymph mites transit from possessing a soft cuticle to forming a tough exoskeleton in adults. In our results, both lncRNAs and mRNAs of some cuticle proteins (cuticle protein 65, cuticle protein 6.4, cuticle protein 63, and cuticle protein 38) were upregulated more in deutonymphs, suggesting that the deutonymph stage is critical in cuticle transition, which is in agreement with the findings in spider mites (Li G. et al., 2021). Another important upregulated gene in deutonymphs was mucin, which is primarily expressed in the peritrophic membrane in the midgut, plays important roles in protecting insects from infection by bacteria, viruses, and other pathogens (Hegedus et al., 2009), and has also been connected to keep cuticle integrity during nymph-adult molting (Zhao et al., 2020). We found that lncRNA LTCONS_00011419 and its target gene mucin 5AC (MUC5AC) were significantly highly expressed in the deutonymph stage than that in the adult stage, emphasizing the significant roles of the deutonymph stage in metamorphosis and pathogen resistance during the development of V. destructor.
Altogether, our present study provides the first integrated analysis of lncRNA-mRNA functional synergistic regulation in the developmental transition of female V. destructor. By depicting lncRNA-mRNA expression profiles and identifying differentially expressed lncRNAs and mRNAs crossing developmental stages, we contributed to gaining a foundational understanding of V. destructor biology. Further evidence from both pathway enrichment and analysis of critical genes indicates that the deutonymph is a key stage in the development of female V. destructor. We hope our results will help in developing more effective treatment techniques for this devastating pest by targeting crucial genes, such as cuticle proteins, in the deutonymph stage.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/ PRJNA777703.
Author Contributions
BH and S-FX conceived and designed the experimental plan. J-LW, R-YH, N-NL, and JT collected the samples. J-LW, C-XZ, and R-YH contributed to reagents, materials, and analysis tools. J-LW, BH, and S-FX wrote the manuscript. All authors approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 31902221), the Modern Agro-Industry Technology Research System (Grant No. CARS-44-KXJ-6), and the Agricultural Science and Technology Innovation Program (Grant No. CAASASTIP-2021-IAR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.842704/full#supplementary-material
Supplementary Figure 1 | Workflow of this study.
Supplementary Figure 2 | The distribution of mRNAs (A) and long non-coding RNAs (lncRNAs) (B) in four developmental stages.
Supplementary Figure 3 | Differentially expressed mRNAs and functional analysis in differently compared libraries. (A) Volcano plot of the mRNAs between protonymph and egg. (B) Volcano plot of the mRNAs between deutonymph and protonymph. (C) Volcano plot of the mRNAs between adult and deutonymph. The left blue points represent significantly decreased mRNAs; gray points represent mRNAs without significant changes; the right red points represent significantly increased mRNAs. (D) The Gene Ontology (GO) terms analysis of the upregulated differentially expressed mRNAs. (E) The GO terms analysis of the downregulated differentially expressed mRNAs.
Supplementary Figure 4 | The enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the differentially expressed mRNA. (A) The KEGG analysis of mRNAs in the protonymph vs. egg comparison group. (B) The KEGG analysis of mRNAs in the deutonymph vs. protonymph comparison group. (C) The KEGG analysis of mRNAs in the adult vs. deutonymph comparison group.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/assembly/GCF_002443255.1
- ^ http://www.geneontology.org/
- ^ http://www.genome.jp/kegg/
References
Arrese, E. L., and Soulages, J. L. (2010). Insect fat body:energy, metabolism, and regulation. Annu. Rev. Entomol. 55, 207–225. doi: 10.1146/annurev-ento-112408-085356
Batista, P. J., and Chang, H. Y. (2013). Long noncoding RNAs:cellular address codes in development and disease. Cell 152, 1298–1307. doi: 10.1016/j.cell.2013.02.012
Boecking, O., and Genersch, E. (2008). Varroosis – the ongoing crisis in bee keeping. J. Verbr. Lebensm. 3, 221–228. doi: 10.1007/s00003-008-0331-y
Chen, D. F., Chen, H. Z., Du, Y., Zhou, D. D., Geng, S. H., Wang, H. P., et al. (2019). Genome-wide identification of long non-coding RNAs and their regulatory networks involved in Apis melliferaligustica response to Nosema ceranae infection. Insects 10:245. doi: 10.3390/insects10080245
Chen, X., Ma, C., Chen, C., Lu, Q., Shi, W., Liu, Z., et al. (2017). Integration of lncRNA-miRNA-mRNA reveals novel insights into oviposition regulation in honey bees. PeerJ 5:e3881. doi: 10.7717/peerj.3881
Chen, X., and Shi, W. (2020). Genome-wide characterization of coding and non-coding RNAs in the ovary of honeybee workers and queens. Apidologie 51, 777–792. doi: 10.1007/s13592-020-00760-7
Cornman, S. R., Schatz, M. C., Johnston, S. J., Chen, Y. P., Pettis, J., Hunt, G., et al. (2010). Genomic survey of the ectoparasitic mite Varroa destructor, a major pest of the honey bee Apis mellifera. BMC Genomics 11:602. doi: 10.1186/1471-2164-11-602
Delas, M. J., and Hannon, G. J. (2017). lncRNAs in development and disease:from functions to mechanisms. Open Biol. 7:170121. doi: 10.1098/rsob.170121
Dermauw, W., and Van Leeuwen, T. (2014). The ABC gene family in arthropods:comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 45, 89–110. doi: 10.1016/j.ibmb.2013.11.001
Dietemann, V., Nazzi, F., Martin, S. J., Anderson, D. L., Locke, B., Delaplane, K. S., et al. (2013). Standard methods for Varroa research. J. Apic. Res. 52:54. doi: 10.3896/Ibra.1.52.1.09
Evans, J. D., and Cook, S. C. (2018). Genetics and physiology of Varroa mites. Curr. Opin. Insect Sci. 26, 130–135. doi: 10.1016/j.cois.2018.02.005
Fatica, A., and Bozzoni, I. (2014). Long non-coding RNAs:new players in cell differentiation and development. Nat. Rev. Genet. 15, 7–21. doi: 10.1038/nrg3606
Feng, K., Liu, J., Wei, P., Ou, S., Wen, X., Shen, G., et al. (2020). lincRNA_Tc13743.2-miR-133-5p-TcGSTm02 regulation pathway mediates cyflumetofen resistance in Tetranychus cinnabarinus. Insect Biochem. Mol. Biol. 123:103413. doi: 10.1016/j.ibmb.2020.103413
Fent, K., Schmid, M., Hettich, T., and Schmid, S. (2020). The neonicotinoid thiacloprid causes transcriptional alteration of genes associated with mitochondria at environmental concentrations in honey bees. Environ. Pollut. 266:115297. doi: 10.1016/j.envpol.2020.115297
Gao, G., and Luo, H. L. (2006). The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 84, 5–14. doi: 10.1139/y05-144
Gardini, A., and Shiekhattar, R. (2015). The many faces of long noncoding RNAs. FEBS J. 282, 1647–1657. doi: 10.1111/febs.13101
Geisler, S., and Coller, J. (2013). RNA in unexpected places:long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 14, 699–712. doi: 10.1038/nrm3679
Gott, R. C., Kunkel, G. R., Zobel, E. S., Lovett, B. R., and Hawthorne, D. J. (2017). Implicating ABC transporters in insecticide resistance:research strategies and a decision framework. J. Econ. Entomol. 110, 667–677. doi: 10.1093/jee/tox041
Guo, R., Chen, D., Xiong, C., Hou, C., Zheng, Y., Fu, Z., et al. (2018a). Identification of long non-coding RNAs in the chalkbrood disease pathogen Ascospheara apis. J. Invertebr. Pathol. 156, 1–5. doi: 10.1016/j.jip.2018.06.001
Guo, R., Chen, D., Xiong, C., Hou, C., Zheng, Y., Fu, Z., et al. (2018b). First identification of long non-coding RNAs in fungal parasite Nosema ceranae. Apidologie 49, 660–670. doi: 10.1007/s13592-018-0593-z
Gupta, A. K., Scully, E. D., Palmer, N. A., Geib, S. M., Sarath, G., Hein, G. L., et al. (2019). Wheat streak mosaic virus alters the transcriptome of its vector, wheat curl mite (Aceria tosichella Keifer), to enhance mite development and population expansion. J. Gen. Virol. 100, 889–910. doi: 10.1099/jgv.0.001256
Guttman, M., Donaghey, J., Carey, B. W., Garber, M., Grenier, J. K., Munson, G., et al. (2011). lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300. doi: 10.1038/nature10398
Hegedus, D., Erlandson, M., Gillott, C., and Toprak, U. (2009). New insights into peritrophic matrix synthesis, architecture, and function. Annu. Rev. Entomol. 54, 285–302. doi: 10.1146/annurev.ento.54.110807.090559
Humann, F. C., Tiberio, G. J., and Hartfelder, K. (2013). Sequence and expression characteristics of long noncoding RNAs in honey bee caste development–potential novel regulators for transgressive ovary size. PLoS One 8:e78915. doi: 10.1371/journal.pone.0078915
Jarroux, J., Morillon, A., and Pinskaya, M. (2017). History, discovery, and classification of lncRNAs. Adv. Exp. Med. Biol. 1008, 1–46. doi: 10.1007/978-981-10-5203-3_1
Jayakodi, M., Jung, J. W., Park, D., Ahn, Y. J., Lee, S. C., Shin, S. Y., et al. (2015). Genome-wide characterization of long intergenic non-coding RNAs (lincRNAs) provides new insight into viral diseases in honey bees Apis cerana and Apis mellifera. BMC Genomics 16:680. doi: 10.1186/s12864-015-1868-7
Kim, W., Miguel-Rojas, C., Wang, J., Townsend, J. P., and Trail, F. (2018). Developmental dynamics of long noncoding RNA expression during sexual fruiting body formation in Fusarium graminearum. mBio 9, e01292–e012118. doi: 10.1128/mBio.01292-18
Kiya, T., Ugajin, A., Kunieda, T., and Kubo, T. (2012). Identification of kakusei, a nuclear non-coding RNA, as an immediate early gene from the honeybee, and its application for neuroethological study. Int. J. Mol. Sci. 13, 15496–15509. doi: 10.3390/ijms131215496
Li, A. Y., Cook, S. C., Sonenshine, D. E., Posada-Florez, F., Noble, N. I. I., Mowery, J., et al. (2019). Insights into the feeding behaviors and biomechanics of Varroa destructor mites on honey bee pupae using electropenetrography and histology. J. Insect Physiol. 119:103950. doi: 10.1016/j.jinsphys.2019.103950
Li, Y., Jin, W., Zhai, B., Chen, Y., Li, G., Zhang, Y., et al. (2021). LncRNAs and their regulatory networks in breast muscle tissue of Chinese Gushi chickens during late postnatal development. BMC Genomics 22:44. doi: 10.1186/s12864-020-07356-6
Li, G., Liu, X., Smagghe, G., Niu, J., and Wang, J. (2021). Genome-wide characterization and identification of long non-coding RNAs during the molting process of a spider mite. Panonychus citri. Int. J. Mol. Sci. 22:6909. doi: 10.3390/ijms22136909
Li, Y. R., Baptista, R. P., and Kissinger, J. C. (2020). Noncoding RNAs in apicomplexan parasites:an update. Trends Parasitol. 36, 835–849. doi: 10.1016/j.pt.2020.07.006
Li, Z., Cai, T., Qin, Y., Zhang, Y., Jin, R., Mao, K., et al. (2020). Transcriptional response of ATP-binding cassette (ABC) transporters to insecticide in the brown planthopper, Nilaparvata lugens (Stal). Insects 11:280. doi: 10.3390/insects11050280
Lin, Z., Liu, Y., Chen, X., Han, C., Wang, W., Ke, Y., et al. (2020). Genome-wide identification of long non-coding RNAs in the gravid ectoparasite Varroa destructor. Front. Genet. 11:575680. doi: 10.3389/fgene.2020.575680
Liu, F., Shi, T., Qi, L., Su, X., Wang, D., Dong, J., et al. (2019). lncRNA profile of Apis mellifera and its possible role in behavioural transition from nurses to foragers. BMC Genomics 20:393. doi: 10.1186/s12864-019-5664-7
Liu, S. H., Xia, Y. D., Zhang, Q., Li, W., Li, R. Y., Liu, Y., et al. (2020). Potential targets for controlling Bactrocera dorsalis using cuticle- and hormone-related genes revealed by a developmental transcriptome analysis. Pest Manag. Sci. 76, 2127–2143. doi: 10.1002/ps.5751
Lu, K., Chen, X., Liu, W., Zhang, Z., Wang, Y., You, K., et al. (2017). Characterization of heat shock protein 70 transcript from Nilaparvata lugens (Stal):its response to temperature and insecticide stresses. Pestic. Biochem. Physiol. 142, 102–110. doi: 10.1016/j.pestbp.2017.01.011
Martin, S. J. (1994). Ontogenesis of the mite Varroa jacobsoni Oud. in worker brood of the honeybee Apis mellifera L. under natural conditions. Exp. Appl. Acarol. 18, 87–100.
McAfee, A., Chan, Q. W. T., Evans, J., and Foster, L. J. (2017). A Varroa destructor protein atlas reveals molecular underpinnings of developmental transitions and sexual differentiation. Mol. Cell. Proteom. 16, 2125–2137. doi: 10.1074/mcp.RA117.000104
Meng, Z., Moroishi, T., and Guan, K. L. (2016). Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. doi: 10.1101/gad.274027.115
Mondet, F., Rau, A., Klopp, C., Rohmer, M., Severac, D., Le Conte, Y., et al. (2018). Transcriptome profiling of the honeybee parasite Varroa destructor provides new biological insights into the mite adult life cycle. BMC Genomics 19:328. doi: 10.1186/s12864-018-4668-z
Nazzi, F., and Le Conte, Y. (2016). Ecology of Varroa destructor, the major ectoparasite of the western honey bee.Apis mellifera. Annu. Rev. Entomol. 61, 417–432. doi: 10.1146/annurev-ento-010715-023731
Necsulea, A., Soumillon, M., Warnefors, M., Liechti, A., Daish, T., Zeller, U., et al. (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640. doi: 10.1038/nature12943
Noël, A., Le Conte, Y., and Mondet, F. (2020). Varroa destructor: how does it harm Apis mellifera honey bees and what can be done about it? Emerg. Top. Life Sci. 4, 45–57. doi: 10.1042/ETLS20190125
Quinn, J. J., and Chang, H. Y. (2016). Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 17, 47–62. doi: 10.1038/nrg.2015.10
Ramsey, S. D., Ochoa, R., Bauchan, G., Gulbronson, C., Mowery, J. D., Cohen, A., et al. (2019). Varroa destructor feeds primarily on honey bee fat body tissue and not hemolymph. PNAS 116, 1792–1801. doi: 10.1073/pnas.1818371116
Rosenkranz, P., Aumeier, P., and Ziegelmann, B. (2010). Biology and control of Varroa destructor. J. Invertebr. Pathol. 103, S96–S119. doi: 10.1016/j.jip.2009.07.016
Sawata, M., Takeuchi, H., and Kubo, T. (2004). Identification and analysis of the minimal promoter activity of a novel noncoding nuclear RNA gene. RNA 10, 1047–1058. doi: 10.1261/rna.5231504
Schor, I. E., Bussotti, G., Males, M., Forneris, M., Viales, R. R., Enright, A. J., et al. (2018). Non-coding RNA expression, function, and variation during Drosophila Embryogenesis. Curr. Biol. 28:e3549. doi: 10.1016/j.cub.2018.09.026
Sun, L., Zhang, Z., Bailey, T. L., Perkins, A. C., Tallack, M. R., Xu, Z., et al. (2012). Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC Bioinformatics 13:331. doi: 10.1186/1471-2105-13-331
Tian, C. B., Li, Y. Y., Huang, J., Chu, W. Q., Wang, Z. Y., and Liu, H. (2020). Comparative transcriptome and proteome analysis of heat acclimation in predatory mite Neoseiulus barkeri. Front. Physiol. 11:426. doi: 10.3389/fphys.2020.00426
Traynor, K. S., Mondet, F., De Miranda, J. R., Techer, M., Kowallik, V., Oddie, M. A. Y., et al. (2020). Varroa destructor:a complex parasite, crippling honey bees worldwide. Trends Parasitol. 36, 592–606. doi: 10.1016/j.pt.2020.04.004
Wang, J. J., Niu, M. H., Zhang, T., Shen, W., and Cao, H. G. (2020). Genome-wide network of lncRNA-mRNA during ovine oocyte development from germinal vesicle to metaphase II in vitro. Front. Physiol. 11:1019. doi: 10.3389/fphys.2020.01019
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi: 10.1016/j.molcel.2011.08.018
Wang, X., Song, X., Glass, C. K., and Rosenfeld, M. G. (2011). The long arm of long noncoding RNAs:roles as sensors regulating gene transcriptional programs. Cold Spring Harb. Perspect. Biol. 3:a003756. doi: 10.1101/cshperspect.a003756
Wedd, L., Kucharski, R., and Maleszka, R. (2016). Differentially methylated obligatory epialleles modulate context-dependent LAM gene expression in the honeybee Apis mellifera. Epigenetics 11, 1–10. doi: 10.1080/15592294.2015.1107695
Yang, L., Wang, Y. W., Lu, Y. Y., Li, B., Chen, K. P., and Li, C. J. (2021). Genome-wide identification and characterization of long non-coding RNAs in Tribolium castaneum. Insect Sci. 28, 1262–1276. doi: 10.1111/1744-7917.12867
Zalewski, K., Zaobidna, E., and Żóltowska, K. (2016). Fatty acid composition of the parasitic mite Varroa destructor and its host the worker prepupae of Apis mellifera. Physiol. Entomol. 41, 31–37.
Zhang, K., Huang, K., Luo, Y., and Li, S. (2014). Identification and functional analysis of long non-coding RNAs in mouse cleavage stage embryonic development based on single cell transcriptome data. BMC Genomics 15:845. doi: 10.1186/1471-2164-15-845
Keywords: Varroa destructor, RNA sequencing, lncRNA, developmental transition, deutonymph
Citation: Wu J-L, Hu R-Y, Li N-N, Tan J, Zhou C-X, Han B and Xu S-F (2022) Integrative Analysis of lncRNA-mRNA Co-expression Provides Novel Insights Into the Regulation of Developmental Transitions in Female Varroa destructor. Front. Ecol. Evol. 10:842704. doi: 10.3389/fevo.2022.842704
Received: 24 December 2021; Accepted: 24 January 2022;
Published: 18 February 2022.
Edited by:
Alessandro Minelli, University of Padua, ItalyReviewed by:
Zheguang Lin, Yangzhou University, ChinaSebastian Kittelmann, Oxford Brookes University, United Kingdom
Copyright © 2022 Wu, Hu, Li, Tan, Zhou, Han and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Han, hanbin@caas.cn; Shu-Fa Xu, xushufa@caas.cn
†These authors have contributed equally to this work and share first authorship