Abstracts
The authors describe two additional cases of Geleophysic dysplasia in siblings, which is a rare autosomal recessive disorder of glycoprotein metabolism whose basic defects remain to be determined.
Os autores descrevem dois novos casos de displasia Geleofísica em irmãos, uma doença autossômica recessiva rara do metabolismo de glicoproteínas cujo defeito básico ainda não foi determinado.
Geleophysic dysplasia - Report on two sibs
Raquel Boy 1, Juan Llerena 2, Márcia Mattos Gonçalves Pimentel 1 and José Carlos Cabral de Almeida 2,3
1 Serviço de Genética Humana, Departamento de Biologia Celular e Genética, Instituto de Biologia, UERJ, Rua São Francisco Xavier, 524 - PHLC, 2o andar, sala 205, Maracanã, 20550-013 Rio de Janeiro, RJ, Brasil. E-mail: pimentel@uerf.br. Send correspondence to M.M.G.P.
2 Centro de Genética Médica, Instituto Fernandes Figueira, FIOCRUZ, Rio de Janeiro, RJ, Brasil.
3 Unidade de Citogenética Humana, Instituto de Biofísica CCFo., UFRJ, Rio de Janeiro, RJ, Brasil.
ABSTRACT
The authors describe two additional cases of Geleophysic dysplasia in siblings, which is a rare autosomal recessive disorder of glycoprotein metabolism whose basic defects remain to be determined.
INTRODUCTION
Geleophysic dysplasia was first described by Spranger et al. (1971). Clinically, the condition belongs to the "acrofacial" group of disorders with short stature and prominent abnormalities in the hands and feet (Wraith et al., 1990). Affected individuals also have a characteristic facial appearance which is described as "happy", and this is the derivation of the conditions name. Other abnormalities associated with the disorder include thick skin, limited joint movement with an unusual tiptoe gait and infiltration of the cardiac valves, trachea and liver with a mucopolysaccharide-like substance. Based on the detection of lysosome-like inclusions in hepatocytes (Wraith et al., 1990), chondrocytes and skin fibroblasts (Pontz et al., 1996), the underlying condition is considered to be a storage defect in the metabolism of glycoproteins.
We report two additional cases of sibs with Geleophysic dysplasia, which support the hypothesis of autosomal recessive inheritance.
CLINICAL REPORT
Patient 1
Patient 1, born in 1989, was the first child of unrelated, young parents. His mother has a history of alcohol abuse and has below-average intelligence. Data for pregnancy were uncertain. He was born at term by normal delivery, weighing 1,800 g. He had one hospital admission during the first three months of life. A delayed psychomotor development was noted - he walked unassisted and talked at 3 years old. By this time, he was also noted to have a systolic murmur, and echocardiography demonstrated perimembranous IVC, persistent left vena cava and dilatation of coronary sinus.
At 5 years 8 months old he was referred to the genetic outpatient clinic for evaluation of short stature and dysmorphic facies. His height at 6 years 2 months old was 95 cm (< 3rd pecentile), OFC 49 cm (50th percentile), and weight 15.200 g (< 5th percentile). Brachycephaly, prominent eyes, ocular hypertelorism, small nose, thickened upper lip with long and flat philtrum and pectus excavatum were observed (Figure 1). An enlarged liver was palpated. Joint mobility was decreased, especially in the hands and elbows. He had very small hands and feet, and a voluminous inguinal hernia was present.
Patient 2
Patient 2, born in 1990, is the sister of patient 1. They have a younger sister in good health. Gestational and neonatal data were also uncertain, except for the normal delivery information. She was evaluated at the genetic outpatient clinic at 4 years for short stature. Her height at 4 years 8 months old was 95 cm (3rd percentile), OFC 48 cm (50th percentile) and weight 15.500 g (< 10th percentile). Her face was very similar to her brothers: hypertelorism, prominent eyes, small nose, and a flat, long philtrum (Figure 2). A cardiac murmur was heard, and an echocardiography dis- closed subaortic ring and aortic coarctation. Short limbs with small hands and feet were also noted. The patients general health has been stable, and both are very pleasant.
- Patients 1 and 2: Ocular hypertelorism, small nose, thickened upper lip with long and flat philtrum, and pectus excavatum.
X-ray findings
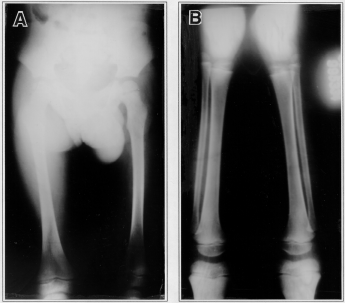
Both patients underwent complete skeletal surveys. Abnormalities were most obvious in patient 1, especially in his hands. Skull roentgenograms showed brachycephaly (Figure 3) and a J-shaped sella turcica. The metacarpals were short, with rounding of proximal ends. Carpal development was somewhat retarded (Figure 4). The long tubular bones were short and broad (Figure 5A,B). No abnormalities were found in the radiographs of the vertebra or pelvis.
Laboratory investigations
Histochemical and ultrastructural studies were performed in the skin biopsy of patient 1, only. Light microscopy was unremarkable and electron microscopy failed to show vacuoles in skin epithelial cells. The patients parents did not allow any other kind of biopsy to be performed.
The patients had normal oligo-saccharide levels in the plasma and urine and thyroid hormone levels (Table I).
Table I - Summary of features found in the two patients.
+, Present; - absent; EM, electron microscopy; IVC, interventricular communication; NS, not studied. Rd - Radiographs or radiologic.
DISCUSSION
The sibs fulfilled the diagnostic criteria for Geleophysic dysplasia. The most notable characteristics were their short limbs, small hands and feet with joint contractures, cardiac abnormalities and characteristic facial appearance. Sibs with the condition have been described in both sexes, which supports the hypothesis of an autosomal-recessive trait (Koiffman et al., 1984; Spranger et al., 1984; Rosser et al., 1995).
Mild developmental delay has also been noted in this condition (Spranger et al., 1971; Koiffman et al., 1984; Shohat et al., 1989; Rosser et al., 1995). Speech delay has been associated with impaired hearing (Rosser et al., 1995), but neither was observed in our cases.
It is interesting to note that patient 2 seems to be less severely affected than her brother. Her hands are not as small and are more mobile, and cardiac involvement is not as severe. Literature reports show that the clinical spectrum can vary with most patients dying in early childhood from severe cardiac disease, and others who are alive and active in the second decade of life (Spranger et al., 1984; Lipson et al., 1987; Wraith et al., 1990).
Serial data from prenatal scans were reported by Rosser et al. (1995), who demonstrated that the growth defects occur relatively late in development. Therefore, prenatal scans are not an accurate detection method for couples at risk.
Some manifestations of Geleophysic dysplasia, such as progressive infiltration of cardiac valves and hepatomegaly, and the discovery of storage vacuoles in chondrocytes and skin fibroblasts, suggest an underlying storage disorder. Liver/skin/iliac crest biopsies of affected individuals have shown patchy changes with numerous lysosome-like vesicular inclusions in hepatocytes / fibroblasts / chondrocytes, but the precise metabolic defect has not been found (Wraith et al., 1990; Pontz et al., 1996).
Like that observed by Wraith et al. (1990), the present case showed no ultrastructural evidence of a lysosomal storage disorder in the skin biopsy. Morphological analyses of other tissues might be helpful in confirming the clinical diagnosis.
RESUMO
Os autores descrevem dois novos casos de displasia Geleofísica em irmãos, uma doença autossômica recessiva rara do metabolismo de glicoproteínas cujo defeito básico ainda não foi determinado.
ACKNOWLEDGMENT
We thank Dr. W. Kattenbach (Departamento de Embriologia e Histologia, Universidade do Estado do Rio de Janeiro) for performing the histochemical and ultrastructural studies.
(Received July 23, 1997)
- Koiffman, C.P., Wajntal, A., Ursich, M.J.M. and Pupo, A.A. (1984). Familial recurrence of geleophysic dysplasia. Am. J. Med. Genet. 19: 483-486.
- Lipson, A.H., Khan, A.E. and Kozlowski, K. (1987). Geleophysic dysplasia-acromicric dysplasia with evidence of glycoprotein storage. Am. J. Med. Genet. 3 (Suppl.): 181-189.
- Pontz, B.F., Stöb, H., Henschke, F., Freisinger, P., Karbowski, A. and Spranger, J. (1996). Clinical and ultrastructural findings in three patients with Geleophysic dysplasia. Am. J. Med. Genet. 63: 50-54.
- Rosser, E.M., Wilkinson, A.R., Hurst, J.A., McGaughran, J.M. and Donnai, D. (1995). Geleophysic dysplasia: report of three affected boys - prenatal ultrasound does not detect recurrence. Am. J. Med. Genet. 58: 217-222.
- Shohat, M., Gruber, H.E. and Payon, R.A (1989). Geleophysic dysplasia: a storage disorder affecting the skin, bone, liver, heart and trachea. J. Med. Genet. 26: 320-325.
- Spranger, J.W., Gilbert, E.F., Tuffli, G.A., Rossiter, F.P. and Opitz, J.M.O. (1971). Geleophysic dwarfism: A "focal" mucopolysaccharidosis? Lancet 2: 97-98.
- Spranger, J.W., Gilbert, E.F., Arya, S., Hoganson, G. and Opitz, J.M. (1984). Geleophysic dysplasia. Am. J. Med. Genet. 19: 487-499.
- Wraith, J.E., Bankier, A., Chow, C.W., Danks, M. and Sardharwalla, I.B. (1990). Geleophysic dysplasia. Am. J. Med. Genet. 35: 153-156.
Publication Dates
-
Publication in this collection
06 Jan 1999 -
Date of issue
Mar 1998
History
-
Received
23 July 1997